publications
Below are selected publications. The complete list of publications is available here.
* indicates equal contribution.
2026
-
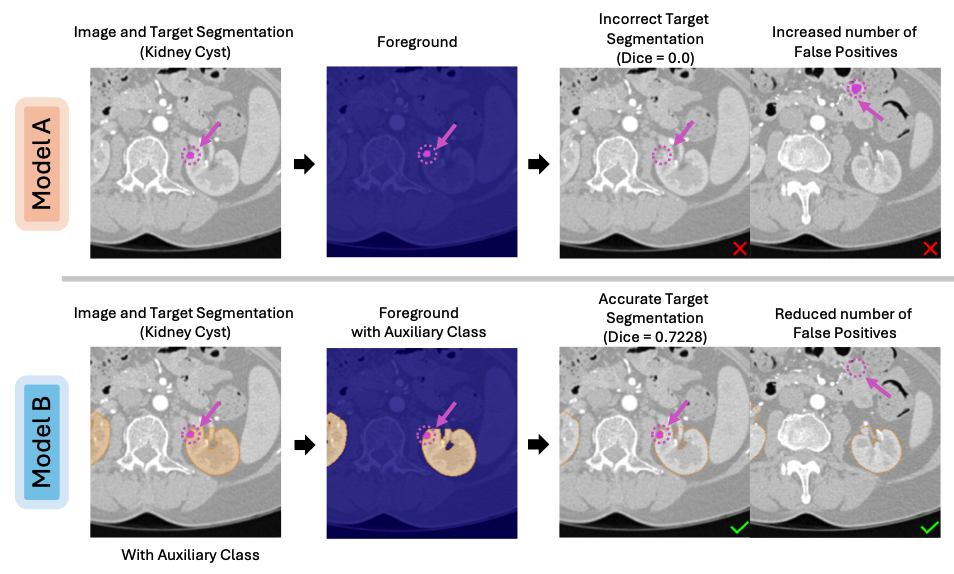 BackSplit: The Importance of Sub-dividing the Background in Biomedical Lesion SegmentationRachit Saluja , Asli Cihangir , Ruining Deng, Johannes C Paetzold , Fengbei Liu , and Mert R SabuncuIn Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition , 2026
BackSplit: The Importance of Sub-dividing the Background in Biomedical Lesion SegmentationRachit Saluja , Asli Cihangir , Ruining Deng, Johannes C Paetzold , Fengbei Liu , and Mert R SabuncuIn Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition , 2026Segmenting small lesions in medical images remains notoriously difficult. Most prior work tackles this challenge by either designing better architectures, loss functions, or data augmentation schemes; and collecting more labeled data. We take a different view, arguing that part of the problem lies in how the background is modeled. Common lesion segmentation collapses all non-lesion pixels into a single "background" class, ignoring the rich anatomical context in which lesions appear. In reality, the background is highly heterogeneous-composed of tissues, organs, and other structures that can now be labeled manually or inferred automatically using existing segmentation models. In this paper, we argue that training with fine-grained labels that sub-divide the background class, which we call BackSplit, is a simple yet powerful paradigm that can offer a significant performance boost without increasing inference costs. From an information theoretic standpoint, we prove that BackSplit increases the expected Fisher Information relative to conventional binary training, leading to tighter asymptotic bounds and more stable optimization. With extensive experiments across multiple datasets and architectures, we empirically show that BackSplit consistently boosts small-lesion segmentation performance, even when auxiliary labels are generated automatically using pretrained segmentation models. Additionally, we demonstrate that auxiliary labels derived from interactive segmentation frameworks exhibit the same beneficial effect, demonstrating its robustness, simplicity, and broad applicability.
@inproceedings{saluja2025backsplit, title = {BackSplit: The Importance of Sub-dividing the Background in Biomedical Lesion Segmentation}, author = {Saluja, Rachit and Cihangir, Asli and Deng, Ruining and Paetzold, Johannes C and Liu, Fengbei and Sabuncu, Mert R}, booktitle = {Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition}, year = {2026}, } -
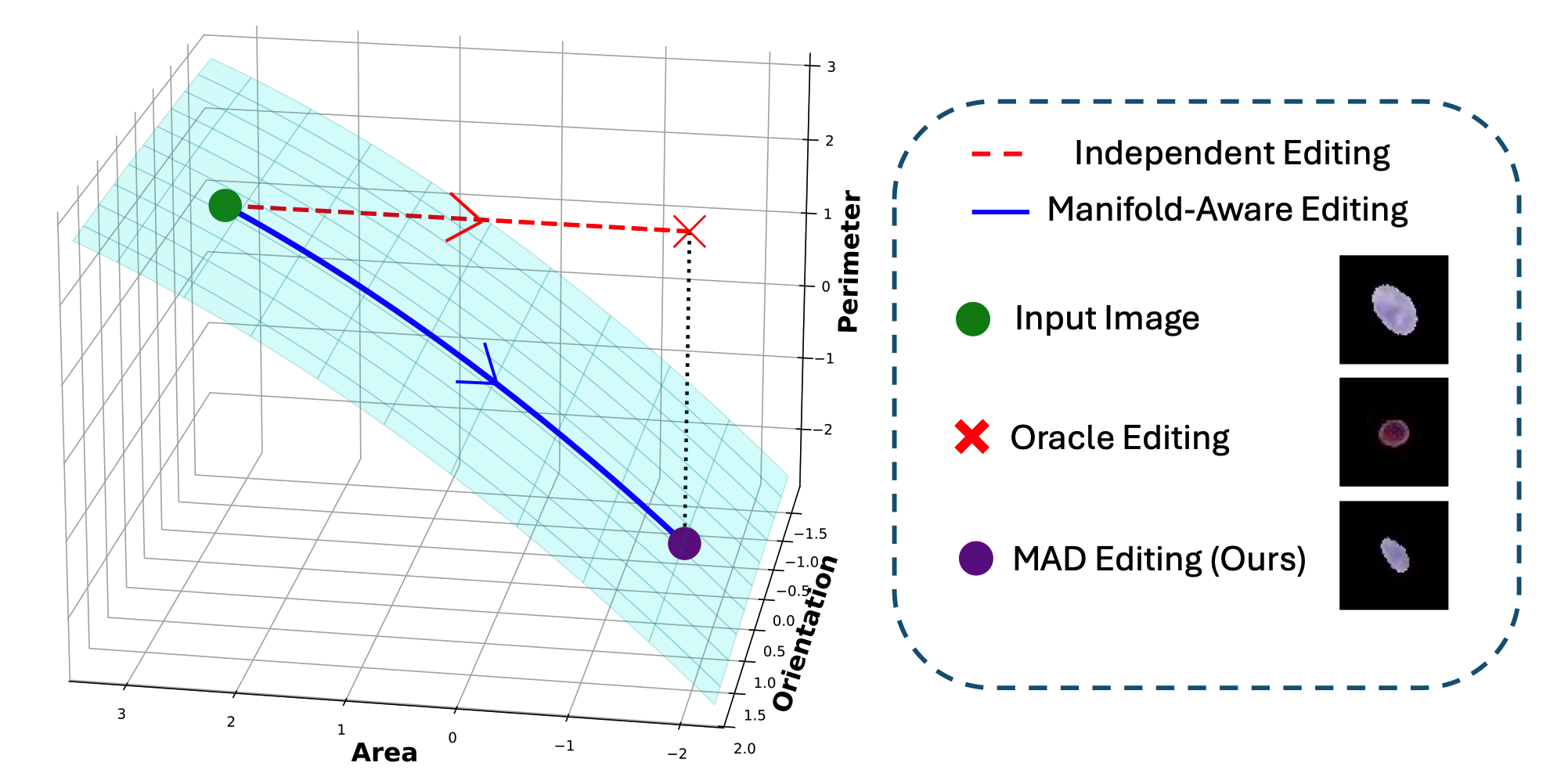 Explainable Pathomics Feature Visualization via Correlation-aware Conditional Feature EditingYuechen Yang , Junlin Guo , Ruining Deng, Junchao Zhu , Zhengyi Lu , Chongyu Qu , Yanfan Zhu , Xingyi Guo , Yu Wang , Shilin Zhao , Haichun Yang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning , 2026Oral Presentation
Explainable Pathomics Feature Visualization via Correlation-aware Conditional Feature EditingYuechen Yang , Junlin Guo , Ruining Deng, Junchao Zhu , Zhengyi Lu , Chongyu Qu , Yanfan Zhu , Xingyi Guo , Yu Wang , Shilin Zhao , Haichun Yang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning , 2026Oral Presentation@inproceedings{yang2026explainable, title = {Explainable Pathomics Feature Visualization via Correlation-aware Conditional Feature Editing}, author = {Yang, Yuechen and Guo, Junlin and Deng, Ruining and Zhu, Junchao and Lu, Zhengyi and Qu, Chongyu and Zhu, Yanfan and Guo, Xingyi and Wang, Yu and Zhao, Shilin and Yang, Haichun and Huo, Yuankai}, booktitle = {International Conference on Medical Imaging with Deep Learning}, year = {2026}, organization = {PMLR}, note = {Oral Presentation} } -
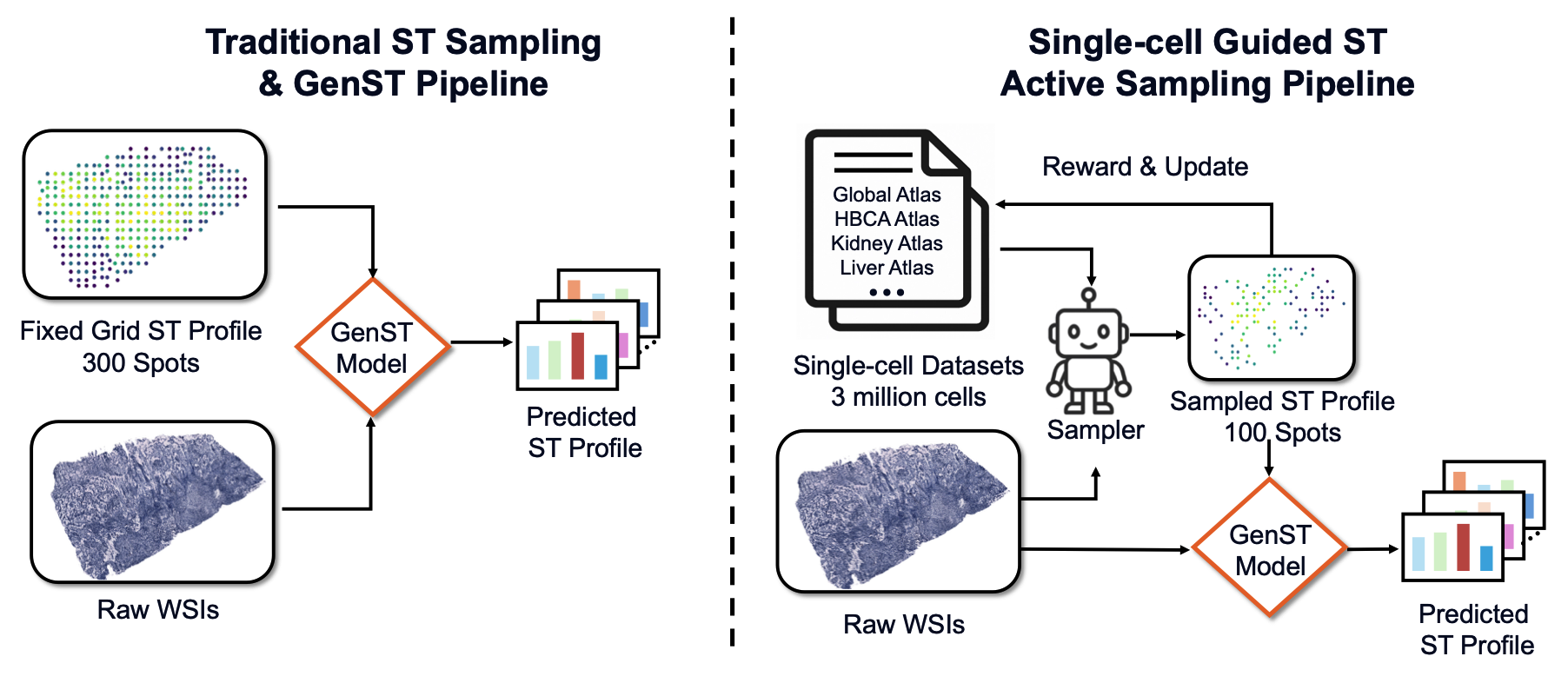 SCR2-ST: Combine Single Cell with Spatial Transcriptomics for Efficient Active Sampling via Reinforcement LearningJunchao Zhu , Ruining Deng, Junlin Guo , Tianyuan Yao , Chongyu Qu , Juming Xiong , Siqi Lu , Zhengyi Lu , Yanfan Zhu , Marilyn Lionts , Yuechen Yang , Yalin Zheng , Yu Wang , Shilin Zhao , Haichun Yang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning , 2026Spotlight Presentation
SCR2-ST: Combine Single Cell with Spatial Transcriptomics for Efficient Active Sampling via Reinforcement LearningJunchao Zhu , Ruining Deng, Junlin Guo , Tianyuan Yao , Chongyu Qu , Juming Xiong , Siqi Lu , Zhengyi Lu , Yanfan Zhu , Marilyn Lionts , Yuechen Yang , Yalin Zheng , Yu Wang , Shilin Zhao , Haichun Yang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning , 2026Spotlight PresentationSpatial transcriptomics (ST) is an emerging technology that enables researchers to investigate the molecular relationships underlying tissue morphology. However, acquiring ST data remains prohibitively expensive, and traditional fixed-grid sampling strategies lead to redundant measurements of morphologically similar or biologically uninformative regions, thus resulting in scarce data that constrain current methods. The well-established single-cell sequencing field, however, could provide rich biological data as an effective auxiliary source to mitigate this limitation. To bridge these gaps, we introduce SCR2-ST, a unified framework that leverages single-cell prior knowledge to guide efficient data acquisition and accurate expression prediction. SCR2-ST integrates a single-cell guided reinforcement learning-based (SCRL) active sampling and a hybrid regression-retrieval prediction network SCR2Net. SCRL combines single-cell foundation model embeddings with spatial density information to construct biologically grounded reward signals, enabling selective acquisition of informative tissue regions under constrained sequencing budgets. SCR2Net then leverages the actively sampled data through a hybrid architecture combining regression-based modeling with retrieval-augmented inference, where a majority cell-type filtering mechanism suppresses noisy matches and retrieved expression profiles serve as soft labels for auxiliary supervision. We evaluated SCR2-ST on three public ST datasets, demonstrating SOTA performance in both sampling efficiency and prediction accuracy, particularly under low-budget scenarios.
@inproceedings{zhu2026scr2, title = {SCR2-ST: Combine Single Cell with Spatial Transcriptomics for Efficient Active Sampling via Reinforcement Learning}, author = {Zhu, Junchao and Deng, Ruining and Guo, Junlin and Yao, Tianyuan and Qu, Chongyu and Xiong, Juming and Lu, Siqi and Lu, Zhengyi and Zhu, Yanfan and Lionts, Marilyn and Yang, Yuechen and Zheng, Yalin and Wang, Yu and Zhao, Shilin and Yang, Haichun and Huo, Yuankai}, booktitle = {International Conference on Medical Imaging with Deep Learning}, year = {2026}, organization = {PMLR}, note = {Spotlight Presentation} } -
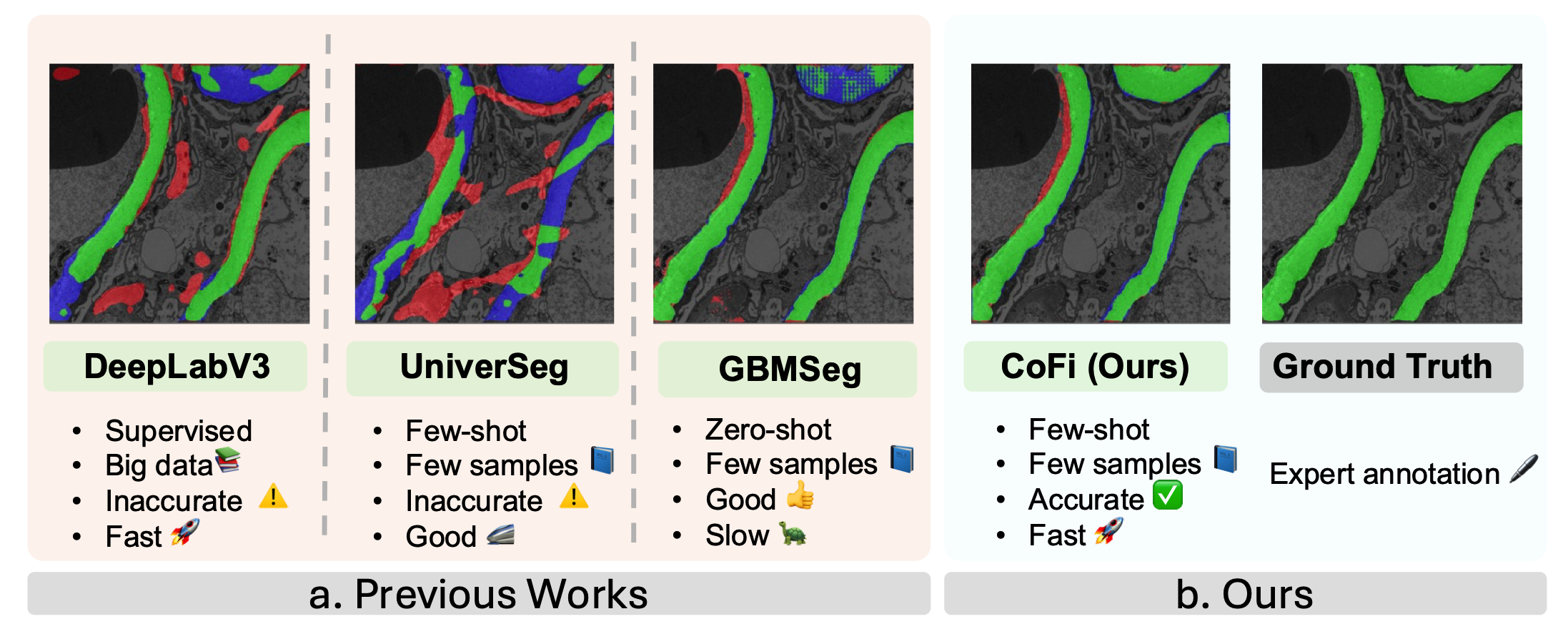 CoFi: a fast coarse-to-fine few-shot pipeline for glomerular basement membrane segmentationHongjin Fang , Daniel Reisenbüchler , Kenji Ikemura , Mert Sabuncu , Yihe Yang , and Ruining DengIn Medical Imaging 2026: Digital and Computational Pathology , 2026
CoFi: a fast coarse-to-fine few-shot pipeline for glomerular basement membrane segmentationHongjin Fang , Daniel Reisenbüchler , Kenji Ikemura , Mert Sabuncu , Yihe Yang , and Ruining DengIn Medical Imaging 2026: Digital and Computational Pathology , 2026Accurate segmentation of the glomerular basement membrane (GBM) in electron microscopy (EM) images is fundamental for quantifying membrane thickness and supporting the diagnosis of various kidney diseases. While supervised deep learning approaches achieve high segmentation accuracy, their reliance on extensive pixel-level annotation renders them impractical for clinical workflows. Few-shot learning can reduce this annotation burden but often struggles to capture the fine structural details necessary for GBM analysis. In this study, we introduce CoFi, a fast and efficient coarse-to-fine few-shot segmentation pipeline designed for GBM delineation in EM images. CoFi first trains a lightweight neural network using only three annotated images to produce an initial coarse segmentation mask. This mask is then automatically processed to generate high-quality point prompts with morphology-aware pruning, which are subsequently used to guide SAM in refining the segmentation. The proposed method achieved exceptional GBM segmentation performance, with a Dice coefficient of 74.54% and an inference speed of 1.9 FPS. We demonstrate that CoFi not only alleviates the annotation and computational burdens associated with conventional methods, but also achieves accurate and reliable segmentation results. The pipeline’s speed and annotation efficiency make it well-suited for research and hold strong potential for clinical applications in renal pathology.
@inproceedings{fang2026cofi, title = {CoFi: a fast coarse-to-fine few-shot pipeline for glomerular basement membrane segmentation}, author = {Fang, Hongjin and Reisenb{\"u}chler, Daniel and Ikemura, Kenji and Sabuncu, Mert and Yang, Yihe and Deng, Ruining}, booktitle = {Medical Imaging 2026: Digital and Computational Pathology}, volume = {13932}, pages = {218--225}, year = {2026}, organization = {SPIE}, } -
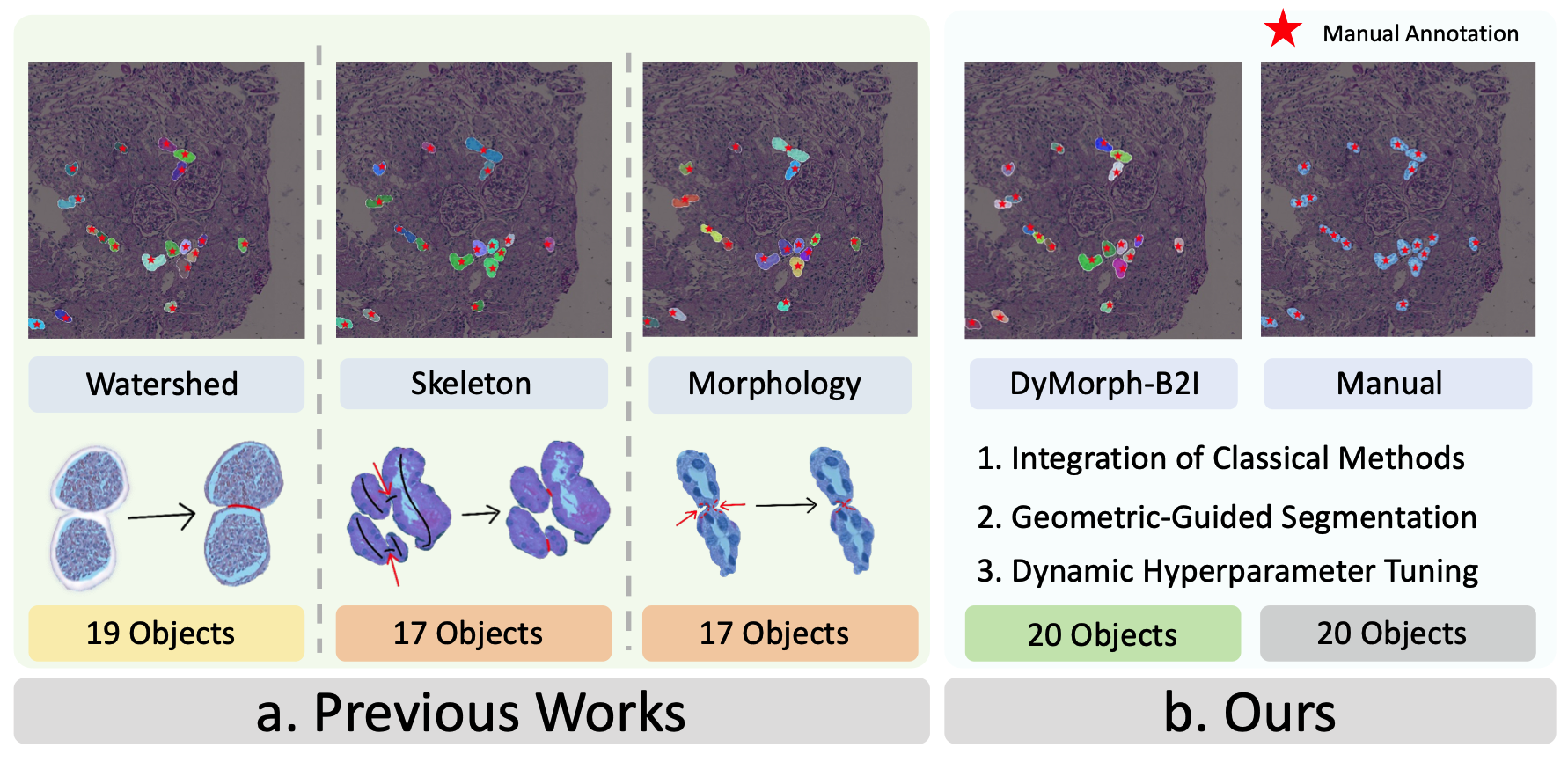 DyMorph-B2I: dynamic and morphology-guided binary-to-instance segmentation for renal pathologyLeiyue Zhao , Yuechen Yang , Yanfan Zhu , Junlin Guo , Haichun Yang , Yuankai Huo , Paul D Simonson , Kenji Ikemura , Mert R Sabuncu , Yihe Yang , and Ruining DengIn Medical Imaging 2026: Digital and Computational Pathology , 2026
DyMorph-B2I: dynamic and morphology-guided binary-to-instance segmentation for renal pathologyLeiyue Zhao , Yuechen Yang , Yanfan Zhu , Junlin Guo , Haichun Yang , Yuankai Huo , Paul D Simonson , Kenji Ikemura , Mert R Sabuncu , Yihe Yang , and Ruining DengIn Medical Imaging 2026: Digital and Computational Pathology , 2026Accurate morphological quantification of renal pathology functional units relies on instance-level segmentation, yet most existing datasets and automated methods provide only binary (semantic) masks, limiting the precision of downstream analyses. Although classical post-processing techniques such as watershed, morphological operations, and skeletonization, are often used to separate semantic masks into instances, their individual effectiveness is constrained by the diverse morphologies and complex connectivity found in renal tissue. In this study, we present DyMorph-B2I, a dynamic, morphology-guided binary-to-instance segmentation pipeline tailored for renal pathology. Our approach integrates watershed, skeletonization, and morphological operations within a unified framework, complemented by adaptive geometric refinement and customizable hyperparameter tuning for each class of functional unit. Through systematic parameter optimization, DyMorph-B2I robustly separates adherent and heterogeneous structures present in binary masks. Experimental results demonstrate that our method outperforms individual classical approaches and naïve combinations, enabling superior instance separation and facilitating more accurate morphometric analysis in renal pathology workflows.
@inproceedings{zhao2026dymorph, title = {DyMorph-B2I: dynamic and morphology-guided binary-to-instance segmentation for renal pathology}, author = {Zhao, Leiyue and Yang, Yuechen and Zhu, Yanfan and Guo, Junlin and Yang, Haichun and Huo, Yuankai and Simonson, Paul D and Ikemura, Kenji and Sabuncu, Mert R and Yang, Yihe and Deng, Ruining}, booktitle = {Medical Imaging 2026: Digital and Computational Pathology}, volume = {13932}, pages = {208--217}, year = {2026}, organization = {SPIE}, } -
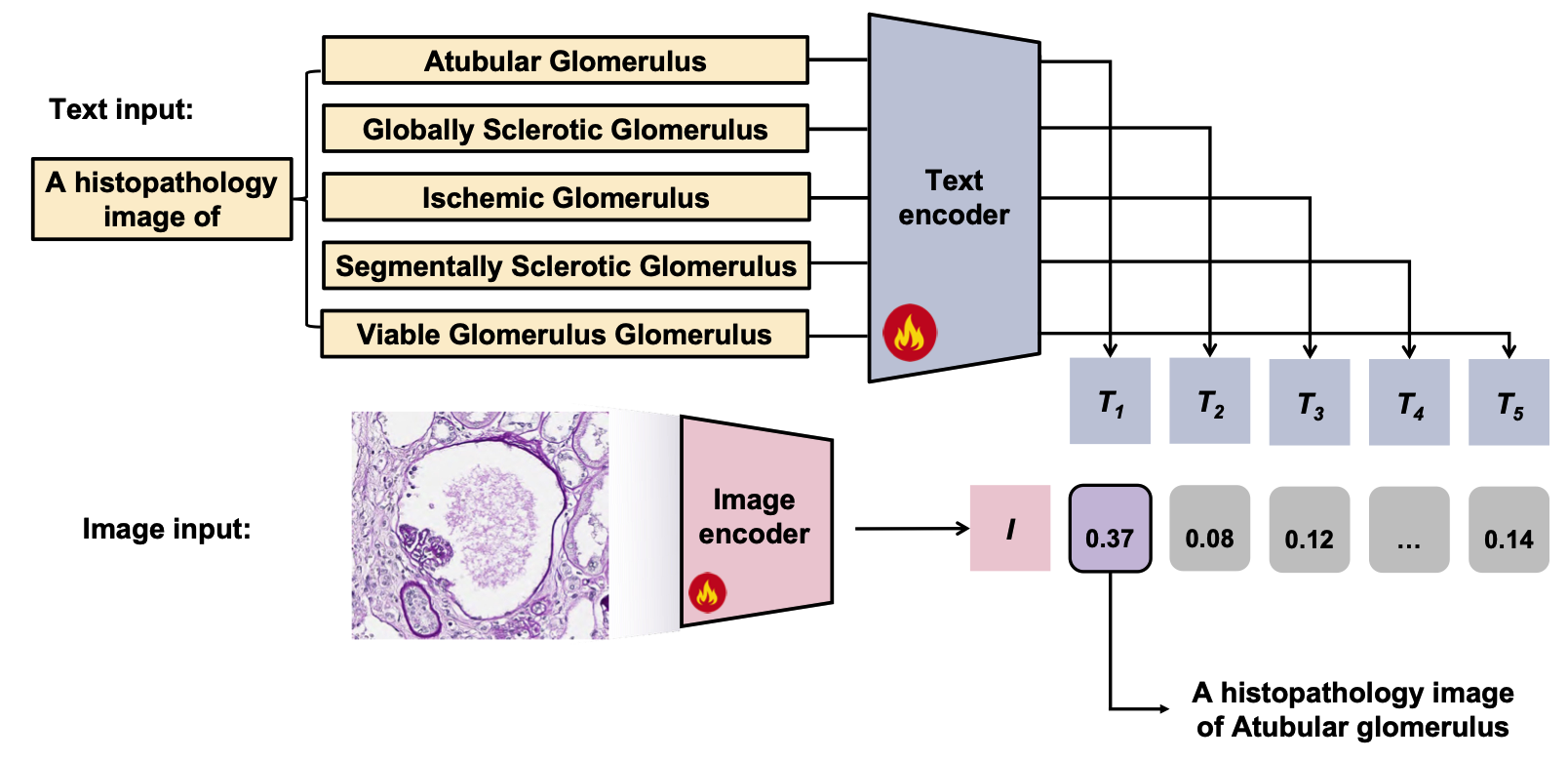 Glo-VLMs: leveraging vision-language models for fine-grained diseased glomerulus classificationZhenhao Guo , Rachit Saluja , Hongjin Fang , Tianyuan Yao , Quan Liu , Yuankai Huo , Benjamin Liechty , David J Pisapia , Kenji Ikemura , Mert R Sabuncu , Yihe Yang , and Ruining DengIn Medical Imaging 2026: Digital and Computational Pathology , 2026
Glo-VLMs: leveraging vision-language models for fine-grained diseased glomerulus classificationZhenhao Guo , Rachit Saluja , Hongjin Fang , Tianyuan Yao , Quan Liu , Yuankai Huo , Benjamin Liechty , David J Pisapia , Kenji Ikemura , Mert R Sabuncu , Yihe Yang , and Ruining DengIn Medical Imaging 2026: Digital and Computational Pathology , 2026Vision-language models (VLMs) have shown considerable potential in digital pathology, yet their effectiveness remains limited for fine-grained, disease-specific classification tasks such as distinguishing between glomerular subtypes. The subtle morphological variations among these subtypes, combined with the difficulty of aligning visual patterns with precise clinical terminology, make automated diagnosis in renal pathology particularly challenging. In this work, we explore how large pretrained VLMs can be effectively adapted to perform fine-grained glomerular classification, even in scenarios where only a small number of labeled examples are available. We introduce Glo-VLMs, a systematic framework designed to explore the adaptation of VLMs to fine-grained glomerular classification in data-constrained settings. Our approach leverages curated pathology images alongside clinical text prompts to facilitate joint image-text representation learning for nuanced renal pathology subtypes. By assessing various VLMs architectures and adaptation strategies under a few-shot learning paradigm, we explore how both the choice of method and the amount of labeled data impact model performance in clinically relevant scenarios. To ensure a fair comparison, we evaluate all models using standardized multi-class metrics. As a result, fine-tuning the VLMs achieved 0.7416 accuracy, 0.9045 macro-AUC, and 0.5277 F1-score with only 8 shots per class, demonstrating that even with highly limited supervision, foundation models can be effectively adapted for fine-grained medical image classification.
@inproceedings{guo2026glo, title = {Glo-VLMs: leveraging vision-language models for fine-grained diseased glomerulus classification}, author = {Guo, Zhenhao and Saluja, Rachit and Fang, Hongjin and Yao, Tianyuan and Liu, Quan and Huo, Yuankai and Liechty, Benjamin and Pisapia, David J and Ikemura, Kenji and Sabuncu, Mert R and Yang, Yihe and Deng, Ruining}, booktitle = {Medical Imaging 2026: Digital and Computational Pathology}, volume = {13932}, pages = {54--61}, year = {2026}, organization = {SPIE}, } -
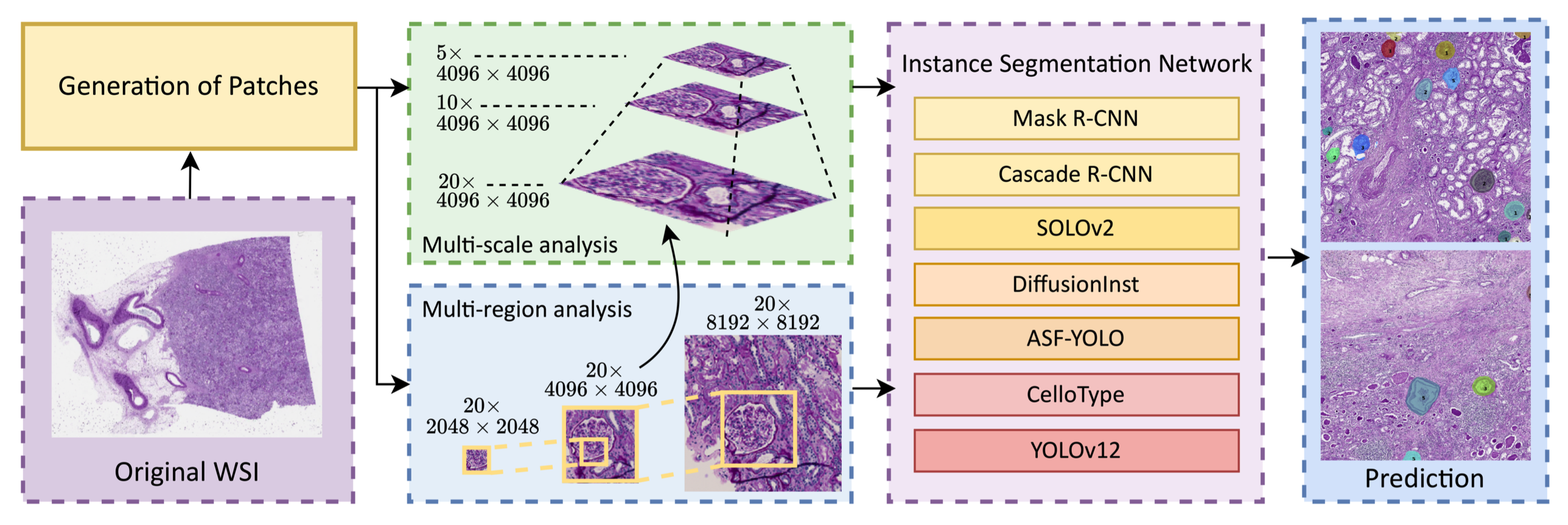 M^3-GloDets: Multi-region and multi-scale analysis of fine-grained diseased glomerular detectionTianyu Shi , Xinzi He , Hongjin Fang , Kenji Ikemura , Mert R Sabuncu , Yihe Yang , and Ruining DengIn Medical Imaging 2026: Digital and Computational Pathology , 2026
M^3-GloDets: Multi-region and multi-scale analysis of fine-grained diseased glomerular detectionTianyu Shi , Xinzi He , Hongjin Fang , Kenji Ikemura , Mert R Sabuncu , Yihe Yang , and Ruining DengIn Medical Imaging 2026: Digital and Computational Pathology , 2026Accurate detection of diseased glomeruli is fundamental to progress in renal pathology and underpins the delivery of reliable clinical diagnoses. Although recent advances in computer vision have produced increasingly sophisticated detection algorithms, the majority of research efforts have focused on normal glomeruli or instances of global sclerosis, leaving the wider spectrum of diseased glomerular subtypes comparatively understudied. The nuanced and highly variable morphological characteristics that define these disease variants frequently elude even the most advanced computational models. Moreover, ongoing debate surrounds the choice of optimal imaging magnifications and region-of-view dimensions for fine-grained glomerular analysis. To bridge these gaps, we present M^3-GloDet, a systematic framework designed to enable thorough evaluation of detection models across a broad continuum of regions, scales, and classes. Within this framework, we evaluate both long-standing benchmark architectures and recently introduced state-of-the-art models, using an experimental design that reflects the diversity of region-of-interest sizes and imaging resolutions encountered in routine digital renal pathology. We found that intermediate patch sizes offered the best balance between context and efficiency, and moderate magnifications enhanced generalization by reducing overfitting. Through systematic comparison on a multi-class diseased glomerular dataset, our aim is to advance the understanding of model strengths and limitations, and to offer actionable insights for the refinement of automated detection strategies and clinical workflows in the digital pathology domain.
@inproceedings{shi2026m, title = {M$^3$-GloDets: Multi-region and multi-scale analysis of fine-grained diseased glomerular detection}, author = {Shi, Tianyu and He, Xinzi and Fang, Hongjin and Ikemura, Kenji and Sabuncu, Mert R and Yang, Yihe and Deng, Ruining}, booktitle = {Medical Imaging 2026: Digital and Computational Pathology}, volume = {13932}, pages = {258--266}, year = {2026}, organization = {SPIE}, }
2025
-
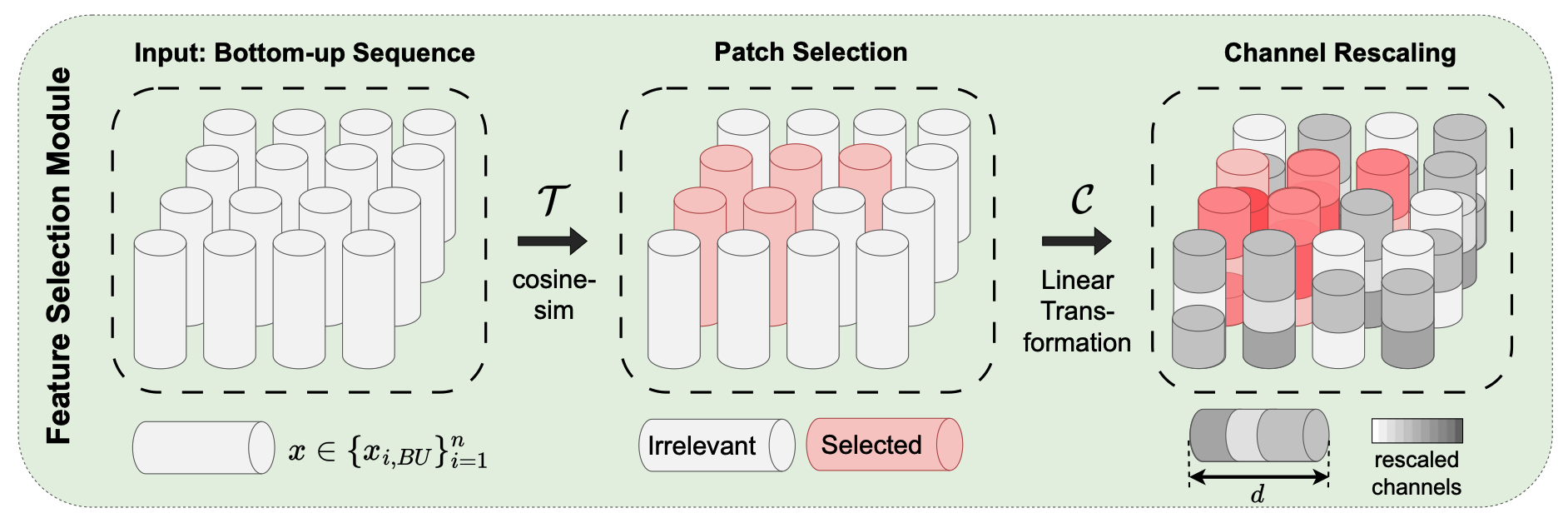 Top-Down Attention-Based Multiple Instance Learning for Whole Slide Image AnalysisDaniel Reisenbüchler , Ruining Deng, Christian Matek , Friedrich Feuerhake , and Dorit MerhofIn International Conference on Medical Image Computing and Computer-Assisted Intervention , 2025
Top-Down Attention-Based Multiple Instance Learning for Whole Slide Image AnalysisDaniel Reisenbüchler , Ruining Deng, Christian Matek , Friedrich Feuerhake , and Dorit MerhofIn International Conference on Medical Image Computing and Computer-Assisted Intervention , 2025Multiple instance learning (MIL) has become the de facto standard approach for whole-slide image analysis in computational pathology (CPath). While instance-wise attention tends to miss correlations between instances, self-attention can capture these interactions, but remains agnostic to the particular task. To address this issue, we introduce Top-Down Attention-based Multiple Instance Learning (TDA-MIL), an architecture that first learns a general representation from the data via self-attention in an initial inference step, then identifies task-relevant instances through a feature selection module, and finally refines these representations by injecting the selected instances back into the attention mechanism for a second inference step. By focusing on task-specific signals, TDA-MIL effectively discerns subtle, yet significant, regions within each slide, leading to more precise classification. Extensive experiments on detecting lymph node metastasis in breast cancer, biomarker screening for microsatellite instability in different organs, and challenging molecular status prediction for HER2 in breast cancer show that TDA-MIL consistently surpasses other MIL baselines, underscoring the effectiveness of our proposed task-relevant refocusing and its broad applicability across CPath tasks.
@inproceedings{reisenbuchler2025top, title = {Top-Down Attention-Based Multiple Instance Learning for Whole Slide Image Analysis}, author = {Reisenb{\"u}chler, Daniel and Deng, Ruining and Matek, Christian and Feuerhake, Friedrich and Merhof, Dorit}, booktitle = {International Conference on Medical Image Computing and Computer-Assisted Intervention}, pages = {651--660}, year = {2025}, organization = {Springer}, } -
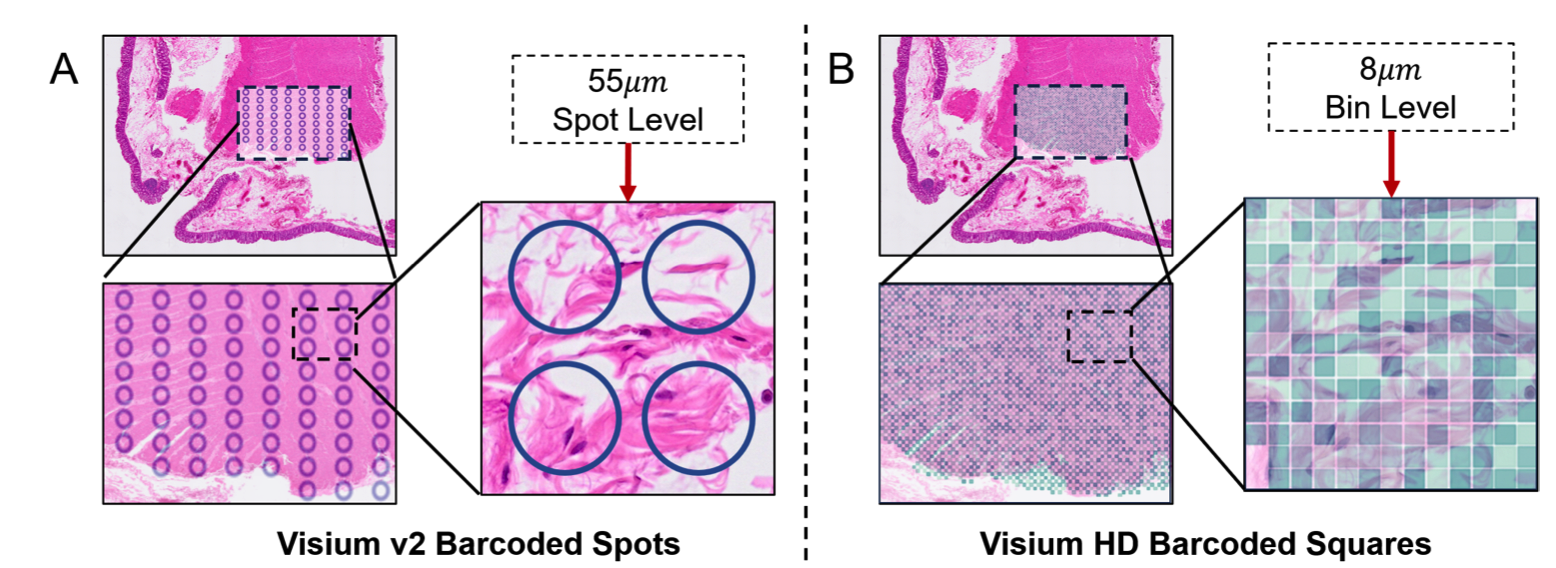 Magnet: Multi-level attention graph network for predicting high-resolution spatial transcriptomicsJunchao Zhu , Ruining Deng, Tianyuan Yao , Juming Xiong , Chongyu Qu , Junlin Guo , Siqi Lu , Yucheng Tang , Daguang Xu , Mengmeng Yin , Yu Wang , Shilin Zhao , Yaohong Wang , Haichun Yang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning , 2025Oral Presentation
Magnet: Multi-level attention graph network for predicting high-resolution spatial transcriptomicsJunchao Zhu , Ruining Deng, Tianyuan Yao , Juming Xiong , Chongyu Qu , Junlin Guo , Siqi Lu , Yucheng Tang , Daguang Xu , Mengmeng Yin , Yu Wang , Shilin Zhao , Yaohong Wang , Haichun Yang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning , 2025Oral PresentationThe rapid development of spatial transcriptomics (ST) offers new opportunities to explore the gene expression patterns within the spatial microenvironment. Current research integrates pathological images to infer gene expression, addressing the high costs and time-consuming processes to generate spatial transcriptomics data. However, as spatial transcriptomics resolution continues to improve, existing methods remain primarily focused on gene expression prediction at low-resolution spot levels. These methods face significant challenges, especially the information bottleneck, when they are applied to high-resolution HD data. To bridge this gap, this paper introduces MagNet, a multi-level attention graph network designed for accurate prediction of high-resolution HD data. MagNet employs cross-attention layers to integrate features from multi-resolution image patches hierarchically and utilizes a GAT-Transformer module to aggregate neighborhood information. By integrating multilevel features, MagNet overcomes the limitations posed by low-resolution inputs in predicting high-resolution gene expression. We systematically evaluated MagNet and existing ST prediction models on both a private spatial transcriptomics dataset and a public dataset at three different resolution levels. The results demonstrate that MagNet achieves state-of-the-art performance at both spot level and high-resolution bin levels, providing a novel methodology and benchmark for future research and applications in high-resolution HD-level spatial transcriptomics.
@inproceedings{zhu2025magnet, title = {Magnet: Multi-level attention graph network for predicting high-resolution spatial transcriptomics}, author = {Zhu, Junchao and Deng, Ruining and Yao, Tianyuan and Xiong, Juming and Qu, Chongyu and Guo, Junlin and Lu, Siqi and Tang, Yucheng and Xu, Daguang and Yin, Mengmeng and Wang, Yu and Zhao, Shilin and Wang, Yaohong and Yang, Haichun and Huo, Yuankai}, booktitle = {International Conference on Medical Imaging with Deep Learning}, year = {2025}, organization = {PMLR}, note = {Oral Presentation} } -
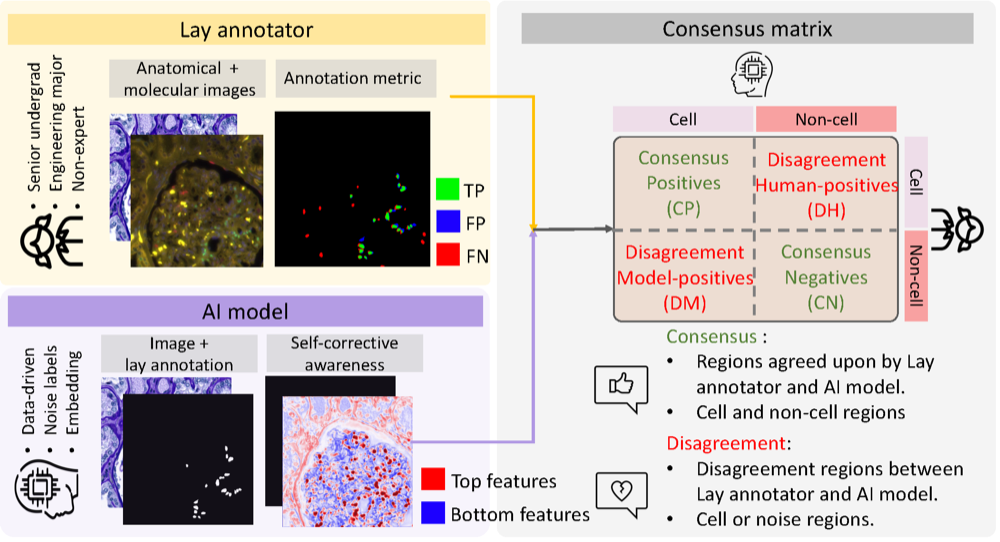 CASC-AI: Consensus-aware Self-corrective Learning for Noise Cell SegmentationRuining Deng, Yihe Yang , David J Pisapia , Benjamin Liechty , Junchao Zhu , Juming Xiong , Junlin Guo , Zhengyi Lu , Jiacheng Wang , Xing Yao , Runxuan Yu , Rendong Zhang , Gaurav Rudravaram , Mengmeng Yin , Pinaki Sarder , Haichun Yang , Yuankai Huo , and Mert R SabuncuIn International Conference on Medical Imaging with Deep Learning , 2025Oral Presentation
CASC-AI: Consensus-aware Self-corrective Learning for Noise Cell SegmentationRuining Deng, Yihe Yang , David J Pisapia , Benjamin Liechty , Junchao Zhu , Juming Xiong , Junlin Guo , Zhengyi Lu , Jiacheng Wang , Xing Yao , Runxuan Yu , Rendong Zhang , Gaurav Rudravaram , Mengmeng Yin , Pinaki Sarder , Haichun Yang , Yuankai Huo , and Mert R SabuncuIn International Conference on Medical Imaging with Deep Learning , 2025Oral PresentationMulti-class cell segmentation in high-resolution gigapixel whole slide images (WSI) is crucial for various clinical applications. However, training such models typically requires labor-intensive, pixel-wise annotations by domain experts. Recent efforts have democratized this process by involving lay annotators without medical expertise. However, conventional non-agent-based approaches struggle to handle annotation noise adaptively, as they lack mechanisms to mitigate false positives (FP) and false negatives (FN) at both the image-feature and pixel levels. In this paper, we propose a consensus-aware self-corrective AI agent that leverages the Consensus Matrix to guide its learning process. The Consensus Matrix defines regions where both the AI and annotators agree on cell and non-cell annotations, which are prioritized with stronger supervision. Conversely, areas of disagreement are adaptively weighted based on their feature similarity to high-confidence agreement regions, with more similar regions receiving greater attention. Additionally, contrastive learning is employed to separate features of noisy regions from those of reliable agreement regions by maximizing their dissimilarity. This paradigm enables the AI to iteratively refine noisy labels, enhancing its robustness. Validated on one real-world lay-annotated cell dataset and two simulated noisy datasets, our method demonstrates improved segmentation performance, effectively correcting FP and FN errors and showcasing its potential for training robust models on noisy datasets. The official implementation and cell annotations are publicly available at https://github.com/ddrrnn123/CASC-AI.
@inproceedings{deng2025casc, title = {CASC-AI: Consensus-aware Self-corrective Learning for Noise Cell Segmentation}, author = {Deng, Ruining and Yang, Yihe and Pisapia, David J and Liechty, Benjamin and Zhu, Junchao and Xiong, Juming and Guo, Junlin and Lu, Zhengyi and Wang, Jiacheng and Yao, Xing and Yu, Runxuan and Zhang, Rendong and Rudravaram, Gaurav and Yin, Mengmeng and Sarder, Pinaki and Yang, Haichun and Huo, Yuankai and Sabuncu, Mert R}, booktitle = {International Conference on Medical Imaging with Deep Learning}, year = {2025}, organization = {PMLR}, note = {Oral Presentation} } -
 ASIGN: An Anatomy-aware Spatial Imputation Graphic Network for 3D Spatial TranscriptomicsJunchao Zhu , Ruining Deng, Tianyuan Yao , Juming Xiong , Chongyu Qu , Junlin Guo , Siqi Lu , Mengmeng Yin , Yu Wang , Shilin Zhao , Haichun Yang , and Yuankai HuoIn Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition , 2025
ASIGN: An Anatomy-aware Spatial Imputation Graphic Network for 3D Spatial TranscriptomicsJunchao Zhu , Ruining Deng, Tianyuan Yao , Juming Xiong , Chongyu Qu , Junlin Guo , Siqi Lu , Mengmeng Yin , Yu Wang , Shilin Zhao , Haichun Yang , and Yuankai HuoIn Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition , 2025Spatial transcriptomics (ST) is an emerging technology that enables medical computer vision scientists to automatically interpret the molecular profiles underlying morphological features. Currently, however, most deep learning-based ST analyses are limited to two-dimensional (2D) sections, which can introduce diagnostic errors due to the heterogeneity of pathological tissues across 3D sections. Expanding ST to three-dimensional (3D) volumes is challenging due to the prohibitive costs; a 2D ST acquisition already costs over 50 times more than whole slide imaging (WSI), and a full 3D volume with 10 sections can be an order of magnitude more expensive. To reduce costs, scientists have attempted to predict ST data directly from WSI without performing actual ST acquisition. However, these methods typically yield unsatisfying results. To address this, we introduce a novel problem setting: 3D ST imputation using 3D WSI histology sections combined with a single 2D ST slide. To do so, we present the Anatomy-aware Spatial Imputation Graph Network (ASIGN) for more precise, yet affordable, 3D ST modeling. The ASIGN architecture extends existing 2D spatial relationships into 3D by leveraging cross-layer overlap and similarity-based expansion. Moreover, a multi-level spatial attention graph network integrates features comprehensively across different data sources. We evaluated ASIGN on three public spatial transcriptomics datasets, with experimental results demonstrating that ASIGN achieves state-of-the-art performance on both 2D and 3D scenarios. Code is available at https://github.com/hrlblab/ASIGN.
@inproceedings{zhu2024asign, title = {ASIGN: An Anatomy-aware Spatial Imputation Graphic Network for 3D Spatial Transcriptomics}, author = {Zhu, Junchao and Deng, Ruining and Yao, Tianyuan and Xiong, Juming and Qu, Chongyu and Guo, Junlin and Lu, Siqi and Yin, Mengmeng and Wang, Yu and Zhao, Shilin and Yang, Haichun and Huo, Yuankai}, booktitle = {Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition}, year = {2025} }
2024
-
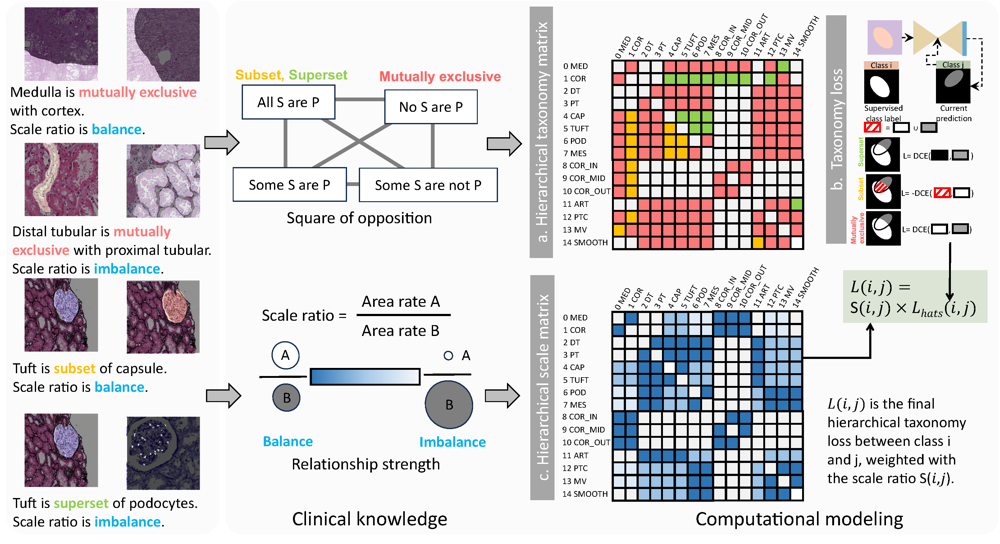 HATs: Hierarchical Adaptive Taxonomy Segmentation for Panoramic Pathology Image AnalysisRuining Deng, Quan Liu , Can Cui , Tianyuan Yao , Juming Xiong , Shunxing Bao , Hao Li , Mengmeng Yin , Yu Wang , Shilin Zhao , Yucheng Tang , Haichun Yang , and Yuankai HuoIn International Conference on Medical Image Computing and Computer-Assisted Intervention , 2024
HATs: Hierarchical Adaptive Taxonomy Segmentation for Panoramic Pathology Image AnalysisRuining Deng, Quan Liu , Can Cui , Tianyuan Yao , Juming Xiong , Shunxing Bao , Hao Li , Mengmeng Yin , Yu Wang , Shilin Zhao , Yucheng Tang , Haichun Yang , and Yuankai HuoIn International Conference on Medical Image Computing and Computer-Assisted Intervention , 2024Multi-class cell segmentation in high-resolution Giga-pixel whole slide images (WSI) is critical for various clinical applications. Training such an AI model typically requires labor-intensive pixel-wise manual annotation from experienced domain experts (e.g., pathologists). Moreover, such annotation is error-prone when differentiating fine-grained cell types (e.g., podocyte and mesangial cells) via the naked human eye. In this study, we assess the feasibility of democratizing pathological AI deployment by only using lay annotators (annotators without medical domain knowledge). The contribution of this paper is threefold: (1) We proposed a molecular-empowered learning scheme for multi-class cell segmentation using partial labels from lay annotators; (2) The proposed method integrated Giga-pixel level molecular-morphology cross-modality registration, molecular-informed annotation, and molecular-oriented segmentation model, so as to achieve significantly superior performance via 3 lay annotators as compared with 2 experienced pathologists; (3) A deep corrective learning (learning with imperfect labels) method is proposed to further improve the segmentation performance using partially annotated noisy data. From the experimental results, our learning method achieved F1 = 0.8496 using molecular-informed annotations from lay annotators, which is better than conventional morphology-based annotations (F1 = 0.7015) from experienced pathologists. Our method democratizes the development of a pathological segmentation deep model to the lay annotator level, which consequently scales up the learning process similar to a non-medical computer vision task. The official implementation and cell annotations are publicly available at https://github.com/hrlblab/MolecularEL.
@inproceedings{deng2024hats, title = {HATs: Hierarchical Adaptive Taxonomy Segmentation for Panoramic Pathology Image Analysis}, author = {Deng, Ruining and Liu, Quan and Cui, Can and Yao, Tianyuan and Xiong, Juming and Bao, Shunxing and Li, Hao and Yin, Mengmeng and Wang, Yu and Zhao, Shilin and Tang, Yucheng and Yang, Haichun and Huo, Yuankai}, booktitle = {International Conference on Medical Image Computing and Computer-Assisted Intervention}, pages = {}, year = {2024}, organization = {Springer}, } -
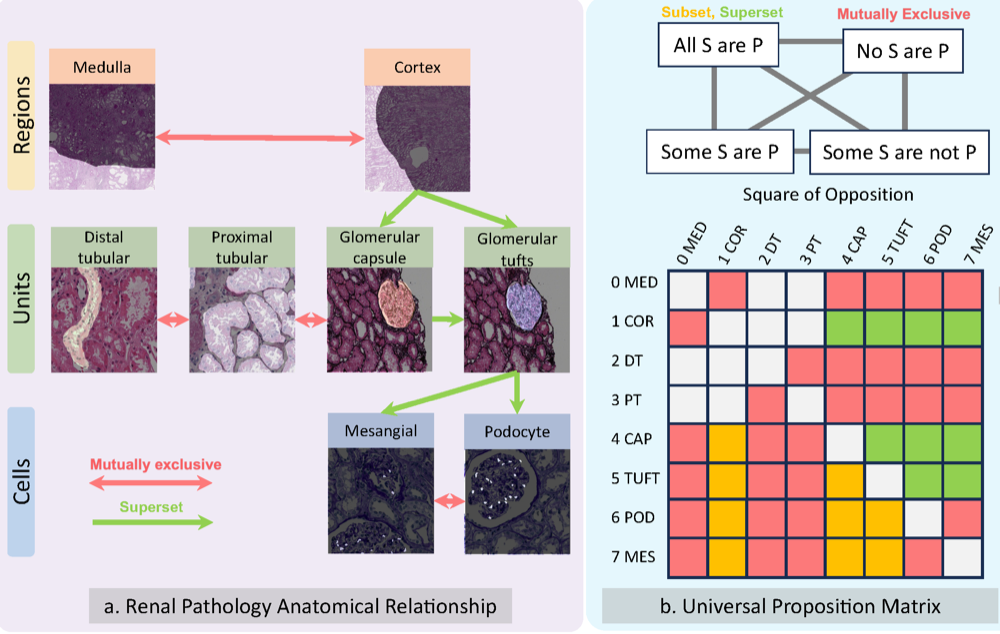 PrPSeg: Universal Proposition Learning for Panoramic Renal Pathology SegmentationRuining Deng, Quan Liu , Can Cui , Tianyuan Yao , Jialin Yue , Juming Xiong , Lining Yu , Yifei Wu , Mengmeng Yin , Yu Wang , Shilin Zhao , Yucheng Tang , Haichun Yang , and Yuankai HuoIn proceedings of the ieee/cvf conference on computer vision and pattern recognition , 2024
PrPSeg: Universal Proposition Learning for Panoramic Renal Pathology SegmentationRuining Deng, Quan Liu , Can Cui , Tianyuan Yao , Jialin Yue , Juming Xiong , Lining Yu , Yifei Wu , Mengmeng Yin , Yu Wang , Shilin Zhao , Yucheng Tang , Haichun Yang , and Yuankai HuoIn proceedings of the ieee/cvf conference on computer vision and pattern recognition , 2024Understanding the anatomy of renal pathology is crucial for advancing disease diagnostics, treatment evaluation, and clinical research. The complex kidney system comprises various components across multiple levels, including regions (cortex, medulla), functional units (glomeruli, tubules), and cells (podocytes, mesangial cells in glomerulus). Prior studies have predominantly overlooked the intricate spatial interrelations among objects from clinical knowledge. In this research, we introduce a novel universal proposition learning approach, called panoramic renal pathology segmentation (PrPSeg), designed to segment comprehensively panoramic structures within kidney by integrating extensive knowledge of kidney anatomy. In this paper, we propose (1) the design of a comprehensive universal proposition matrix for renal pathology, facilitating the incorporation of classification and spatial relationships into the segmentation process; (2) a token-based dynamic head single network architecture, with the improvement of the partial label image segmentation and capability for future data enlargement; and (3) an anatomy loss function, quantifying the inter-object relationships across the kidney.
@inproceedings{deng2024prpseg, title = {PrPSeg: Universal Proposition Learning for Panoramic Renal Pathology Segmentation}, author = {Deng, Ruining and Liu, Quan and Cui, Can and Yao, Tianyuan and Yue, Jialin and Xiong, Juming and Yu, Lining and Wu, Yifei and Yin, Mengmeng and Wang, Yu and Zhao, Shilin and Tang, Yucheng and Yang, Haichun and Huo, Yuankai}, booktitle = {proceedings of the ieee/cvf conference on computer vision and pattern recognition}, pages = {11736--11746}, year = {2024}, } -
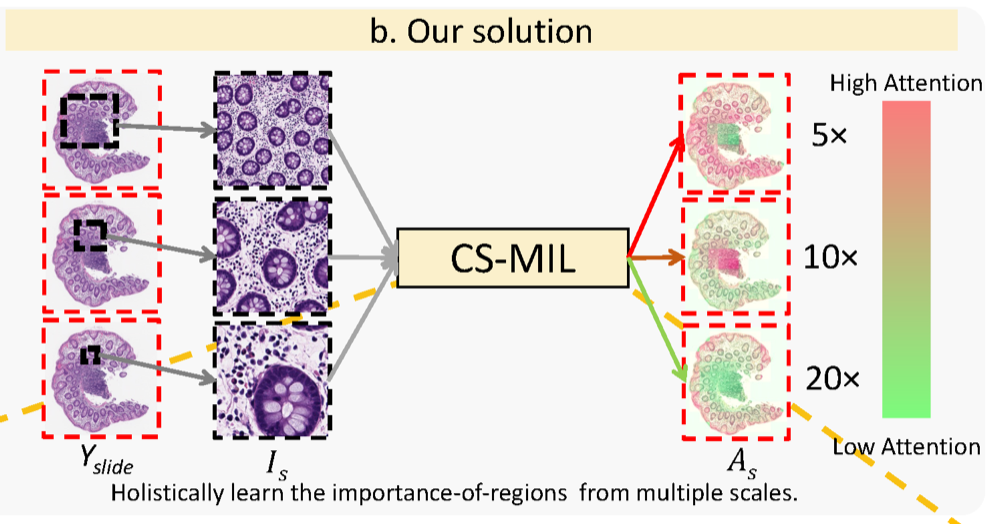 Cross-scale multi-instance learning for pathological image diagnosisRuining Deng, Can Cui , Lucas W Remedios , Shunxing Bao , R Michael Womick , Sophie Chiron , Jia Li , Joseph T Roland , Ken S Lau , Qi Liu , Keith T Wilson , Yaohong Wang , Lori A Coburn , Bennett A Landman , and Yuankai HuoMedical Image Analysis, 2024
Cross-scale multi-instance learning for pathological image diagnosisRuining Deng, Can Cui , Lucas W Remedios , Shunxing Bao , R Michael Womick , Sophie Chiron , Jia Li , Joseph T Roland , Ken S Lau , Qi Liu , Keith T Wilson , Yaohong Wang , Lori A Coburn , Bennett A Landman , and Yuankai HuoMedical Image Analysis, 2024Analyzing high resolution whole slide images (WSIs) with regard to information across multiple scales poses a significant challenge in digital pathology. Multi-instance learning (MIL) is a common solution for working with high resolution images by classifying bags of objects (i.e. sets of smaller image patches). However, such processing is typically performed at a single scale (e.g., 20 magnification) of WSIs, disregarding the vital inter-scale information that is key to diagnoses by human pathologists. In this study, we propose a novel cross-scale MIL algorithm to explicitly aggregate inter-scale relationships into a single MIL network for pathological image diagnosis. The contribution of this paper is three-fold: (1) A novel cross-scale MIL (CS-MIL) algorithm that integrates the multi-scale information and the inter-scale relationships is proposed; (2) A toy dataset with scale-specific morphological features is created and released to examine and visualize differential cross-scale attention; (3) Superior performance on both in-house and public datasets is demonstrated by our simple cross-scale MIL strategy. The official implementation is publicly available at https://github.com/hrlblab/CS-MIL.
@article{deng2024cross, title = {Cross-scale multi-instance learning for pathological image diagnosis}, author = {Deng, Ruining and Cui, Can and Remedios, Lucas W and Bao, Shunxing and Womick, R Michael and Chiron, Sophie and Li, Jia and Roland, Joseph T and Lau, Ken S and Liu, Qi and Wilson, Keith T and Wang, Yaohong and Coburn, Lori A and Landman, Bennett A and Huo, Yuankai}, journal = {Medical Image Analysis}, pages = {103124}, year = {2024}, publisher = {Elsevier}, } -
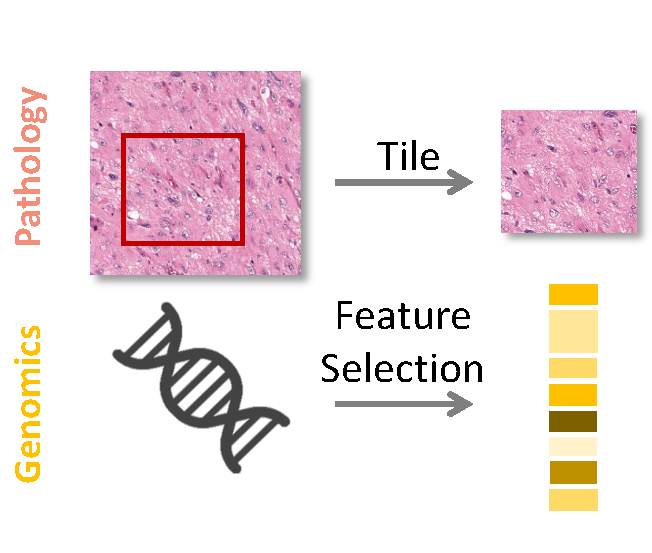 Cross-modality attention-based multimodal fusion for non-small cell lung cancer (NSCLC) patient survival predictionRuining Deng, Nazim Shaikh , Gareth Shannon , and Yao NieIn Medical Imaging 2024: Digital and Computational Pathology , 2024Best Paper Finalist
Cross-modality attention-based multimodal fusion for non-small cell lung cancer (NSCLC) patient survival predictionRuining Deng, Nazim Shaikh , Gareth Shannon , and Yao NieIn Medical Imaging 2024: Digital and Computational Pathology , 2024Best Paper FinalistCancer prognosis and survival outcome predictions are crucial for therapeutic response estimation and for stratifying patients into various treatment groups. Medical domains concerned with cancer prognosis are abundant with multiple modalities, including pathological image data and non-image data such as genomic information. To date, multimodal learning has shown potential to enhance clinical prediction model performance by extracting and aggregating information from different modalities of the same subject. This approach could outperform single modality learning, thus improving computer-aided diagnosis and prognosis in numerous medical applications. In this work, we propose a cross-modality attention-based multimodal fusion pipeline designed to integrate modality-specific knowledge for patient survival prediction in non-small cell lung cancer (NSCLC). Instead of merely concatenating or summing up the features from different modalities, our method gauges the importance of each modality for feature fusion with cross-modality relationship when infusing the multimodal features. Compared with single modality, which achieved c-index of 0.5772 and 0.5885 using solely tissue image data or RNA-seq data, respectively, the proposed fusion approach achieved c-index 0.6587 in our experiment, showcasing the capability of assimilating modality-specific knowledge from varied modalities.
@inproceedings{deng2024crost, title = {Cross-modality attention-based multimodal fusion for non-small cell lung cancer (NSCLC) patient survival prediction}, author = {Deng, Ruining and Shaikh, Nazim and Shannon, Gareth and Nie, Yao}, booktitle = {Medical Imaging 2024: Digital and Computational Pathology}, volume = {12933}, pages = {46--50}, year = {2024}, organization = {SPIE}, note = {Best Paper Finalist} } -
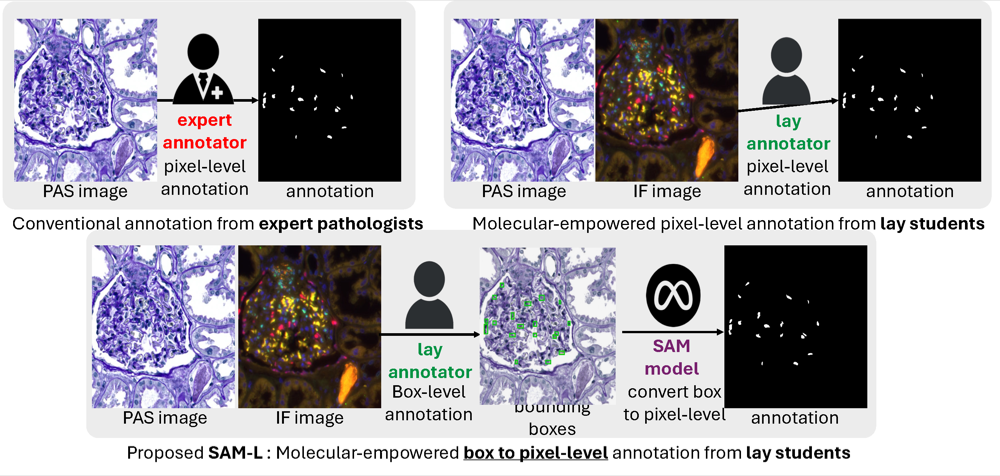 Leverage weekly annotation to pixel-wise annotation via zero-shot segment anything model for molecular-empowered learningXueyuan Li* , Ruining Deng*, Yucheng Tang , Shunxing Bao , Haichun Yang , and Yuankai HuoIn Medical Imaging 2024: Digital and Computational Pathology , 2024Best Paper Finalist
Leverage weekly annotation to pixel-wise annotation via zero-shot segment anything model for molecular-empowered learningXueyuan Li* , Ruining Deng*, Yucheng Tang , Shunxing Bao , Haichun Yang , and Yuankai HuoIn Medical Imaging 2024: Digital and Computational Pathology , 2024Best Paper FinalistPrecise identification of multiple cell classes in high-resolution Giga-pixel whole slide imaging (WSI) is critical for various clinical scenarios. Building an AI model for this purpose typically requires pixel-level annotations, which are often unscalable and must be done by skilled domain experts (e.g., pathologists). However, these annotations can be prone to errors, especially when distinguishing between intricate cell types (e.g., podocytes and mesangial cells) using only visual inspection. Interestingly, a recent study showed that lay annotators, when using extra immunofluorescence (IF) images for reference (referred to as molecular-empowered learning), can sometimes outperform domain experts in labeling. Despite this, the resource-intensive task of manual delineation remains a necessity during the annotation process. In this paper, we explore the potential of bypassing pixel-level delineation by employing the recent segment anything model (SAM) on weak box annotation in a zero-shot learning approach. Specifically, we harness SAM’s ability to produce pixel-level annotations from box annotations and utilize these SAM-generated labels to train a segmentation model. Our findings show that the proposed SAM-assisted molecular-empowered learning (SAM-L) can diminish the labeling efforts for lay annotators by only requiring weak box annotations. This is achieved without compromising annotation accuracy or the performance of the deep learning-based segmentation. This research represents a significant advancement in democratizing the annotation process for training pathological image segmentation, relying solely on non-expert annotators.
@inproceedings{li2024leverage, title = {Leverage weekly annotation to pixel-wise annotation via zero-shot segment anything model for molecular-empowered learning}, author = {Li*, Xueyuan and Deng*, Ruining and Tang, Yucheng and Bao, Shunxing and Yang, Haichun and Huo, Yuankai}, booktitle = {Medical Imaging 2024: Digital and Computational Pathology}, volume = {12933}, pages = {133--139}, year = {2024}, organization = {SPIE}, note = {Best Paper Finalist} } -
 Multi-scale multi-site renal microvascular structures segmentation for whole slide imaging in renal pathologyFranklin Hu* , Ruining Deng*, Shunxing Bao , Haichun Yang , and Yuankai HuoIn Medical Imaging 2024: Digital and Computational Pathology , 2024
Multi-scale multi-site renal microvascular structures segmentation for whole slide imaging in renal pathologyFranklin Hu* , Ruining Deng*, Shunxing Bao , Haichun Yang , and Yuankai HuoIn Medical Imaging 2024: Digital and Computational Pathology , 2024Segmenting microvascular structures, such as arterioles, venules, and capillaries, from human kidney whole slide images (WSI) in renal pathology has garnered significant interest. The current manual segmentation approach is laborious and impractical for large-scale digital pathology images. To address this, deep learning-based methods have emerged for automatic segmentation. However, a gap exists in current deep learning segmentation methods, as they are typically designed and limited by using single-site single-scale data for training. In this paper, we introduce a novel single dynamic network method (Omni-Seg), which harnesses multi-site multi-scale training data, utilizing partially labeled images where only one tissue type is labeled per training image for microvascular structure segmentation. We train a single deep network using images from two datasets, HuBMAP and NEPTUNE, with different scales (40×, 20×, 10×, 5×). Our experimental results demonstrate that our approach achieves higher Dice Similarity Coefficient (DSC) and Intersection over Union (IoU) scores. This proposed method empowers renal pathologists with a computational tool for quantitatively assessing renal microvascular structures.
@inproceedings{hu2024multi, title = {Multi-scale multi-site renal microvascular structures segmentation for whole slide imaging in renal pathology}, author = {Hu*, Franklin and Deng*, Ruining and Bao, Shunxing and Yang, Haichun and Huo, Yuankai}, booktitle = {Medical Imaging 2024: Digital and Computational Pathology}, volume = {12933}, pages = {310--316}, year = {2024}, organization = {SPIE}, }
2023
-
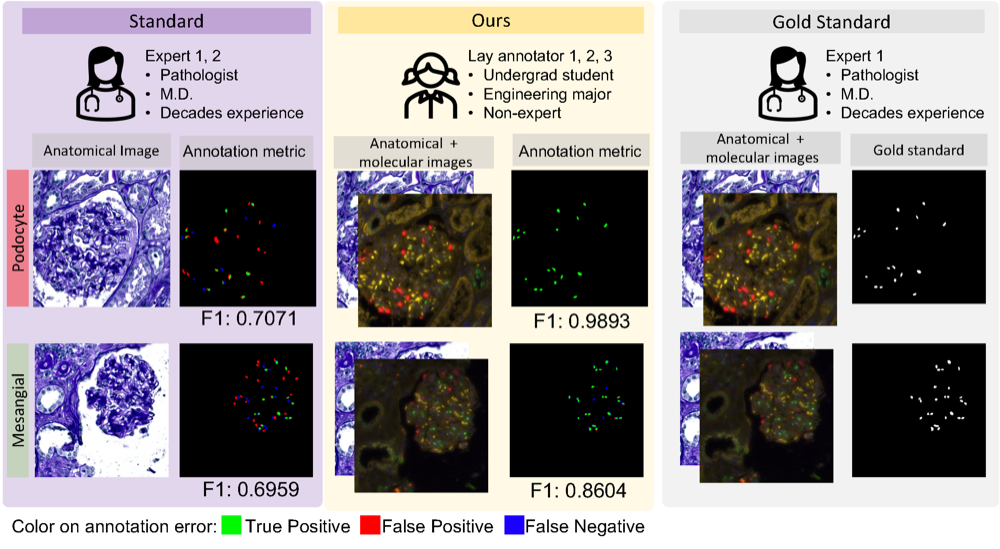 Democratizing pathological image segmentation with lay annotators via molecular-empowered learningRuining Deng, Yanwei Li , Peize Li , Jiacheng Wang , Lucas W Remedios , Saydolimkhon Agzamkhodjaev , Zuhayr Asad , Quan Liu , Can Cui , Yaohong Wang , Yihan Wang , Yucheng Tang , Haichun Yang , and Yuankai HuoIn International Conference on Medical Image Computing and Computer-Assisted Intervention , 2023
Democratizing pathological image segmentation with lay annotators via molecular-empowered learningRuining Deng, Yanwei Li , Peize Li , Jiacheng Wang , Lucas W Remedios , Saydolimkhon Agzamkhodjaev , Zuhayr Asad , Quan Liu , Can Cui , Yaohong Wang , Yihan Wang , Yucheng Tang , Haichun Yang , and Yuankai HuoIn International Conference on Medical Image Computing and Computer-Assisted Intervention , 2023Multi-class cell segmentation in high-resolution Giga-pixel whole slide images (WSI) is critical for various clinical applications. Training such an AI model typically requires labor-intensive pixel-wise manual annotation from experienced domain experts (e.g., pathologists). Moreover, such annotation is error-prone when differentiating fine-grained cell types (e.g., podocyte and mesangial cells) via the naked human eye. In this study, we assess the feasibility of democratizing pathological AI deployment by only using lay annotators (annotators without medical domain knowledge). The contribution of this paper is threefold: (1) We proposed a molecular-empowered learning scheme for multi-class cell segmentation using partial labels from lay annotators; (2) The proposed method integrated Giga-pixel level molecular-morphology cross-modality registration, molecular-informed annotation, and molecular-oriented segmentation model, so as to achieve significantly superior performance via 3 lay annotators as compared with 2 experienced pathologists; (3) A deep corrective learning (learning with imperfect labels) method is proposed to further improve the segmentation performance using partially annotated noisy data. From the experimental results, our learning method achieved F1 = 0.8496 using molecular-informed annotations from lay annotators, which is better than conventional morphology-based annotations (F1 = 0.7015) from experienced pathologists. Our method democratizes the development of a pathological segmentation deep model to the lay annotator level, which consequently scales up the learning process similar to a non-medical computer vision task. The official implementation and cell annotations are publicly available at https://github.com/hrlblab/MolecularEL.
@inproceedings{deng2023democratizing, title = {Democratizing pathological image segmentation with lay annotators via molecular-empowered learning}, author = {Deng, Ruining and Li, Yanwei and Li, Peize and Wang, Jiacheng and Remedios, Lucas W and Agzamkhodjaev, Saydolimkhon and Asad, Zuhayr and Liu, Quan and Cui, Can and Wang, Yaohong and Wang, Yihan and Tang, Yucheng and Yang, Haichun and Huo, Yuankai}, booktitle = {International Conference on Medical Image Computing and Computer-Assisted Intervention}, pages = {497--507}, year = {2023}, organization = {Springer}, } -
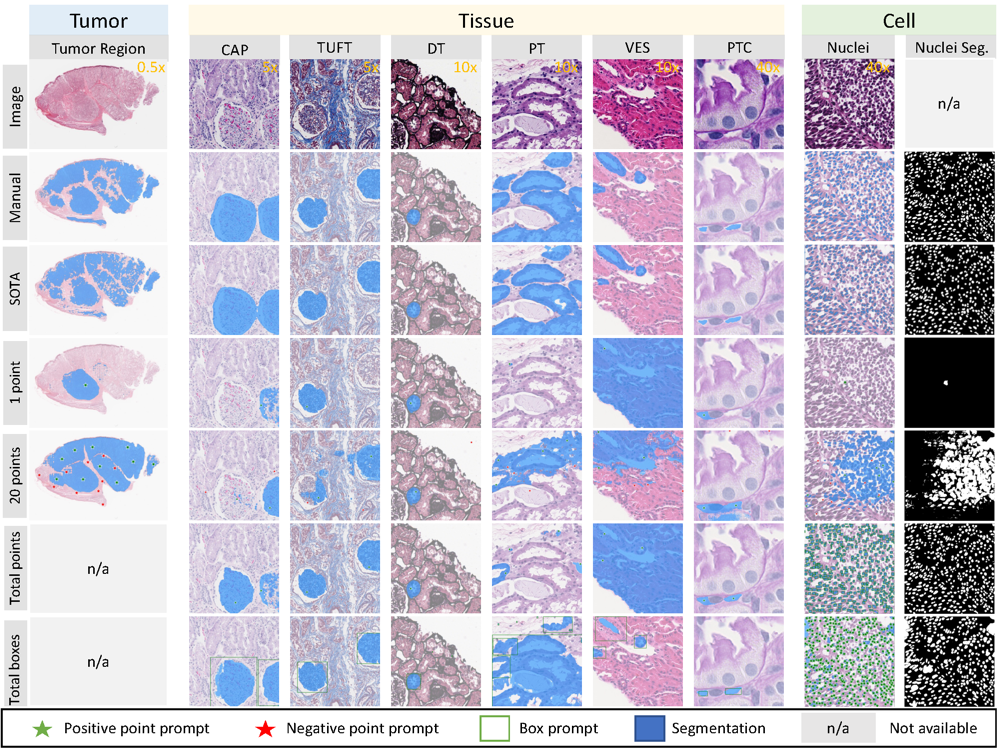 Segment anything model (sam) for digital pathology: Assess zero-shot segmentation on whole slide imagingRuining Deng*, Can Cui* , Quan Liu* , Tianyuan Yao , Lucas W Remedios , Shunxing Bao , Bennett A Landman , Lee E Wheless , Lori A Coburn , Keith T Wilson , Yaohong Wang , Shilin Zhao , Agnes B Fogo , Haichun Yang , Yucheng Tang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning Short Paper Track, 2023
Segment anything model (sam) for digital pathology: Assess zero-shot segmentation on whole slide imagingRuining Deng*, Can Cui* , Quan Liu* , Tianyuan Yao , Lucas W Remedios , Shunxing Bao , Bennett A Landman , Lee E Wheless , Lori A Coburn , Keith T Wilson , Yaohong Wang , Shilin Zhao , Agnes B Fogo , Haichun Yang , Yucheng Tang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning Short Paper Track, 2023The segment anything model (SAM) was released as a foundation model for image segmentation. The promptable segmentation model was trained by over 1 billion masks on 11M licensed and privacy-respecting images. The model supports zero-shot image segmentation with various segmentation prompts (e.g., points, boxes, masks). It makes the SAM attractive for medical image analysis, especially for digital pathology where the training data are rare. In this study, we evaluate the zero-shot segmentation performance of SAM model on representative segmentation tasks on whole slide imaging (WSI), including (1) tumor segmentation, (2) non-tumor tissue segmentation, (3) cell nuclei segmentation. Core Results: The results suggest that the zero-shot SAM model achieves remarkable segmentation performance for large connected objects. However, it does not consistently achieve satisfying performance for dense instance object segmentation, even with 20 prompts (clicks/boxes) on each image. We also summarized the identified limitations for digital pathology: (1) image resolution, (2) multiple scales, (3) prompt selection, and (4) model fine-tuning. In the future, the few-shot fine-tuning with images from downstream pathological segmentation tasks might help the model to achieve better performance in dense object segmentation.
@article{deng2023segment, title = {Segment anything model (sam) for digital pathology: Assess zero-shot segmentation on whole slide imaging}, author = {Deng*, Ruining and Cui*, Can and Liu*, Quan and Yao, Tianyuan and Remedios, Lucas W and Bao, Shunxing and Landman, Bennett A and Wheless, Lee E and Coburn, Lori A and Wilson, Keith T and Wang, Yaohong and Zhao, Shilin and Fogo, Agnes B and Yang, Haichun and Tang, Yucheng and Huo, Yuankai}, journal = {In International Conference on Medical Imaging with Deep Learning Short Paper Track}, year = {2023}, publisher = {PMLR}, } -
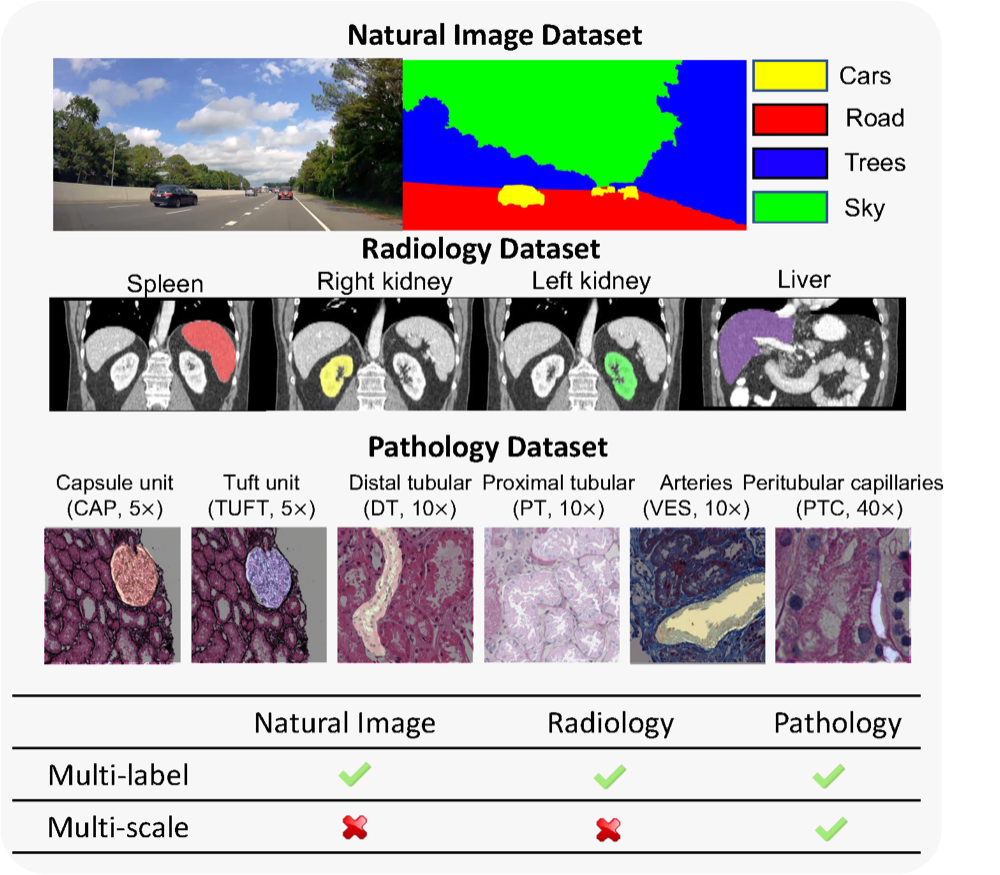 Omni-seg: A scale-aware dynamic network for renal pathological image segmentationRuining Deng, Quan Liu , Can Cui , Tianyuan Yao , Jun Long , Zuhayr Asad , R Michael Womick , Zheyu Zhu , Agnes B Fogo , Shilin Zhao , Haichun Yang , and Yuankai HuoIEEE Transactions on Biomedical Engineering, 2023Best Paper Runner-up
Omni-seg: A scale-aware dynamic network for renal pathological image segmentationRuining Deng, Quan Liu , Can Cui , Tianyuan Yao , Jun Long , Zuhayr Asad , R Michael Womick , Zheyu Zhu , Agnes B Fogo , Shilin Zhao , Haichun Yang , and Yuankai HuoIEEE Transactions on Biomedical Engineering, 2023Best Paper Runner-upComprehensive semantic segmentation on renal pathological images is challenging due to the heterogeneous scales of the objects. For example, on a whole slide image (WSI), the cross-sectional areas of glomeruli can be 64 times larger than that of the peritubular capillaries, making it impractical to segment both objects on the same patch, at the same scale. To handle this scaling issue, prior studies have typically trained multiple segmentation networks in order to match the optimal pixel resolution of heterogeneous tissue types. This multi-network solution is resource-intensive and fails to model the spatial relationship between tissue types. In this article, we propose the Omni-Seg network, a scale-aware dynamic neural network that achieves multi-object (six tissue types) and multi-scale (5× to 40× scale) pathological image segmentation via a single neural network. The contribution of this article is three-fold: (1) a novel scale-aware controller is proposed to generalize the dynamic neural network from single-scale to multi-scale; (2) semi-supervised consistency regularization of pseudo-labels is introduced to model the inter-scale correlation of unannotated tissue types into a single end-to-end learning paradigm; and (3) superior scale-aware generalization is evidenced by directly applying a model trained on human kidney images to mouse kidney images, without retraining. By learning from 150,000 human pathological image patches from six tissue types at three different resolutions, our approach achieved superior segmentation performance according to human visual assessment and evaluation of image-omics (i.e., spatial transcriptomics).
@article{deng2023omni, title = {Omni-seg: A scale-aware dynamic network for renal pathological image segmentation}, author = {Deng, Ruining and Liu, Quan and Cui, Can and Yao, Tianyuan and Long, Jun and Asad, Zuhayr and Womick, R Michael and Zhu, Zheyu and Fogo, Agnes B and Zhao, Shilin and Yang, Haichun and Huo, Yuankai}, journal = {IEEE Transactions on Biomedical Engineering}, year = {2023}, publisher = {IEEE}, note = {Best Paper Runner-up} } -
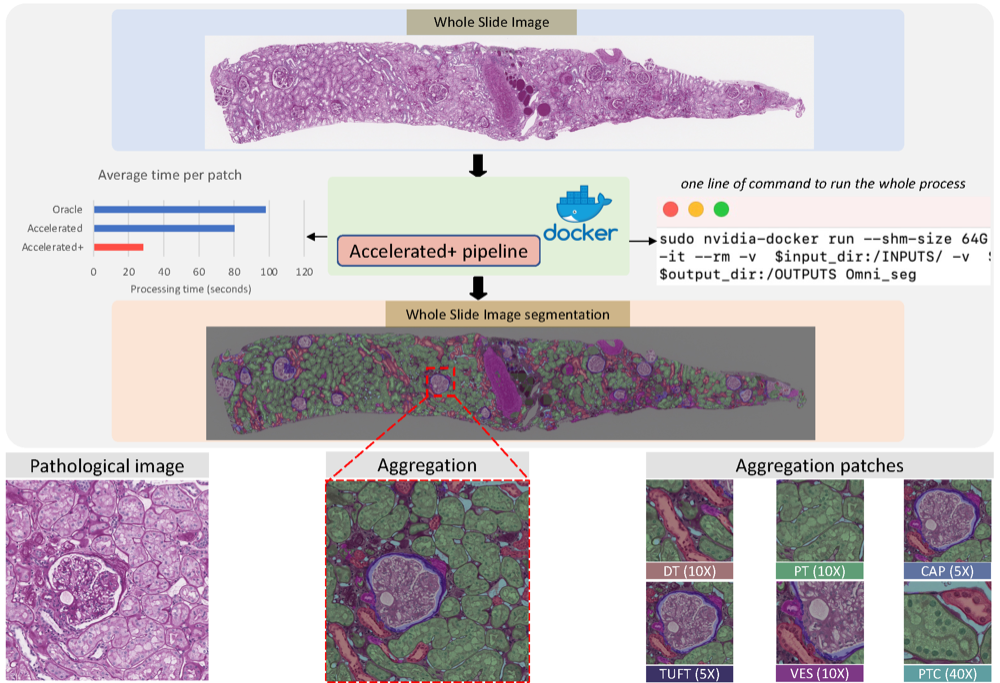 An accelerated pipeline for multi-label renal pathology image segmentation at the whole slide image levelHaoju Leng* , Ruining Deng*, Zuhayr Asad , R Michael Womick , Haichun Yang , Lipeng Wan , and Yuankai HuoIn Medical Imaging 2023: Digital and Computational Pathology , 2023
An accelerated pipeline for multi-label renal pathology image segmentation at the whole slide image levelHaoju Leng* , Ruining Deng*, Zuhayr Asad , R Michael Womick , Haichun Yang , Lipeng Wan , and Yuankai HuoIn Medical Imaging 2023: Digital and Computational Pathology , 2023Deep-learning techniques have been used widely to alleviate the labour-intensive and time-consuming manual annotation required for pixel-level tissue characterization. Our previous study introduced an efficient single dynamic network - Omni-Seg - that achieved multi-class multi-scale pathological segmentation with less computational complexity. However, the patch-wise segmentation paradigm still applies to Omni-Seg, and the pipeline is time-consuming when providing segmentation for Whole Slide Images (WSIs). In this paper, we propose an enhanced version of the Omni-Seg pipeline in order to reduce the repetitive computing processes and utilize a GPU to accelerate the model’s prediction for both better model performance and faster speed. Our proposed method’s innovative contribution is two-fold: (1) a Docker is released for an end-to-end slide-wise multi-tissue segmentation for WSIs; and (2) the pipeline is deployed on a GPU to accelerate the prediction, achieving better segmentation quality in less time. The proposed accelerated implementation reduced the average processing time (at the testing stage) on a standard needle biopsy WSI from 2.3 hours to 22 minutes, using 35 WSIs from the Kidney Tissue Atlas (KPMP) Datasets. The source code and the Docker have been made publicly available at https://github.com/ddrrnn123/Omni-Seg.
@inproceedings{leng2023accelerated, title = {An accelerated pipeline for multi-label renal pathology image segmentation at the whole slide image level}, author = {Leng*, Haoju and Deng*, Ruining and Asad, Zuhayr and Womick, R Michael and Yang, Haichun and Wan, Lipeng and Huo, Yuankai}, booktitle = {Medical Imaging 2023: Digital and Computational Pathology}, volume = {12471}, pages = {174--179}, year = {2023}, organization = {SPIE}, } -
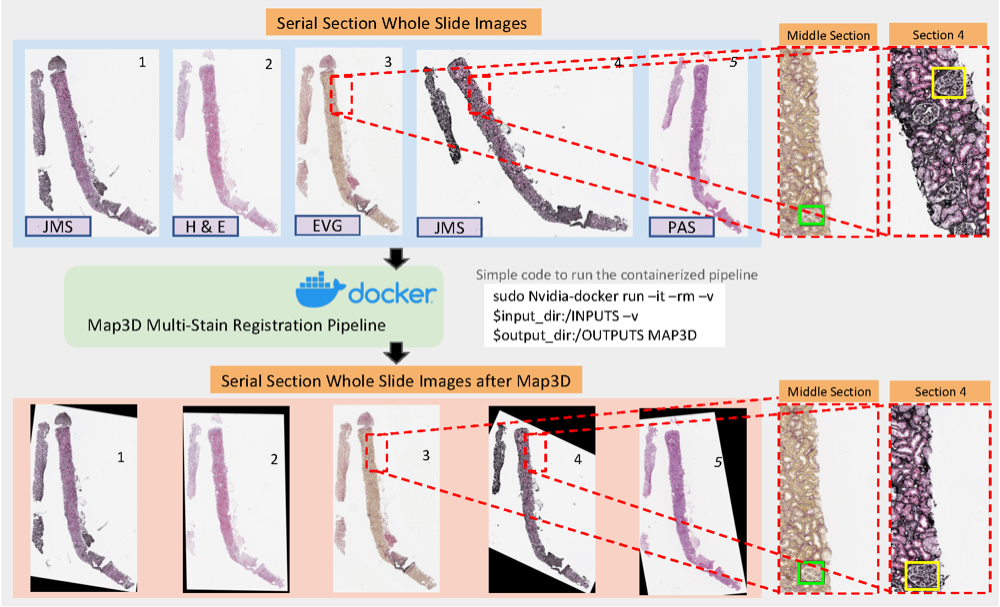 An end-to-end pipeline for 3D slide-wise multi-stain renal pathology registrationPeize Li* , Ruining Deng*, and Yuankai HuoIn Medical Imaging 2023: Digital and Computational Pathology , 2023
An end-to-end pipeline for 3D slide-wise multi-stain renal pathology registrationPeize Li* , Ruining Deng*, and Yuankai HuoIn Medical Imaging 2023: Digital and Computational Pathology , 2023Tissue examination and quantification in a 3D context on serial section whole slide images (WSIs) were laborintensive and time-consuming tasks. Our previous study proposed a novel registration-based method (Map3D) to automatically align WSIs to the same physical space, reducing the human efforts of screening serial sections from WSIs. However, the registration performance of our Map3D method was only evaluated on single-stain WSIs with large-scale kidney tissue samples. In this paper, we provide a Docker for an end-to-end 3D slide-wise registration pipeline on needle biopsy serial sections in a multi-stain paradigm. The contribution of this study is three-fold: (1) We release a containerized Docker for an end-to-end multi-stain WSI registration. (2) We prove that the Map3D pipeline is capable of sectional registration from multi-stain WSI. (3) We verify that the Map3D pipeline can also be applied to needle biopsy tissue samples.
@inproceedings{li2023end, title = {An end-to-end pipeline for 3D slide-wise multi-stain renal pathology registration}, author = {Li*, Peize and Deng*, Ruining and Huo, Yuankai}, booktitle = {Medical Imaging 2023: Digital and Computational Pathology}, volume = {12471}, pages = {96--101}, year = {2023}, organization = {SPIE}, }
2022
-
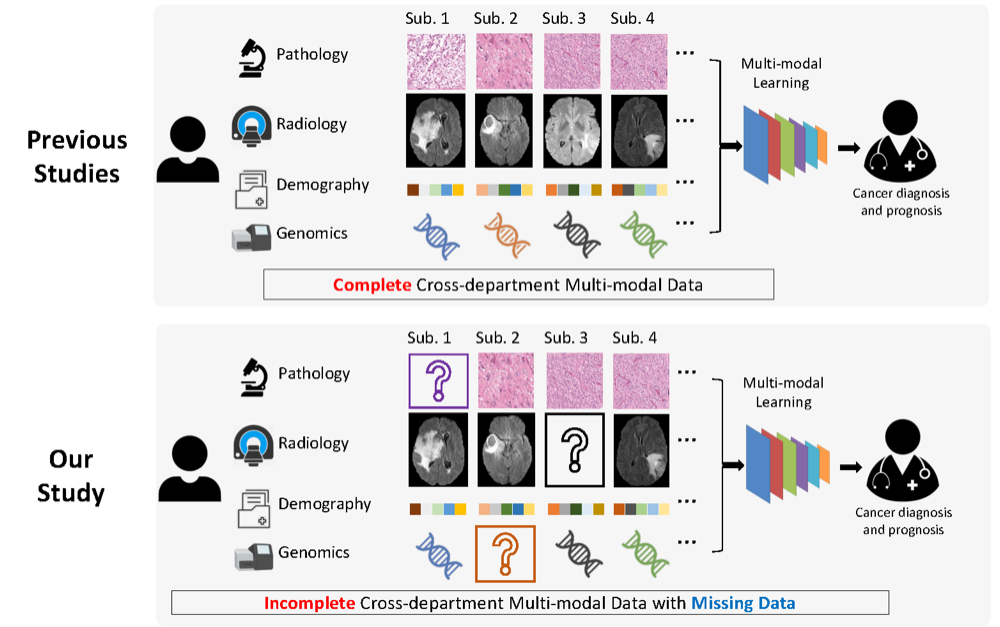 Survival prediction of brain cancer with incomplete radiology, pathology, genomic, and demographic dataCan Cui , Han Liu , Quan Liu , Ruining Deng, Zuhayr Asad , Yaohong Wang , Shilin Zhao , Haichun Yang , Bennett A Landman , and Yuankai HuoIn International Conference on Medical Image Computing and Computer-Assisted Intervention , 2022
Survival prediction of brain cancer with incomplete radiology, pathology, genomic, and demographic dataCan Cui , Han Liu , Quan Liu , Ruining Deng, Zuhayr Asad , Yaohong Wang , Shilin Zhao , Haichun Yang , Bennett A Landman , and Yuankai HuoIn International Conference on Medical Image Computing and Computer-Assisted Intervention , 2022Integrating cross-department multi-modal data (e.g., radiology, pathology, genomic, and demographic data) is ubiquitous in brain cancer diagnosis and survival prediction. To date, such an integration is typically conducted by human physicians (and panels of experts), which can be subjective and semi-quantitative. Recent advances in multi-modal deep learning, however, have opened a door to leverage such a process in a more objective and quantitative manner. Unfortunately, the prior arts of using four modalities on brain cancer survival prediction are limited by a “complete modalities” setting (i.e., with all modalities available). Thus, there are still open questions on how to effectively predict brain cancer survival from incomplete radiology, pathology, genomic, and demographic data (e.g., one or more modalities might not be collected for a patient). For instance, should we use both complete and incomplete data, and more importantly, how do we use such data? To answer the preceding questions, we generalize the multi-modal learning on cross-department multi-modal data to a missing data setting. Our contribution is three-fold: 1) We introduce a multi-modal learning with missing data (MMD) pipeline with competitive performance and less hardware consumption; 2) We extend multi-modal learning on radiology, pathology, genomic, and demographic data into missing data scenarios; 3) A large-scale public dataset (with 962 patients) is collected to systematically evaluate glioma tumor survival prediction using four modalities. The proposed method improved the C-index of survival prediction from 0.7624 to 0.8053.
@inproceedings{cui2022survival, title = {Survival prediction of brain cancer with incomplete radiology, pathology, genomic, and demographic data}, author = {Cui, Can and Liu, Han and Liu, Quan and Deng, Ruining and Asad, Zuhayr and Wang, Yaohong and Zhao, Shilin and Yang, Haichun and Landman, Bennett A and Huo, Yuankai}, booktitle = {International Conference on Medical Image Computing and Computer-Assisted Intervention}, pages = {626--635}, year = {2022}, organization = {Springer}, } -
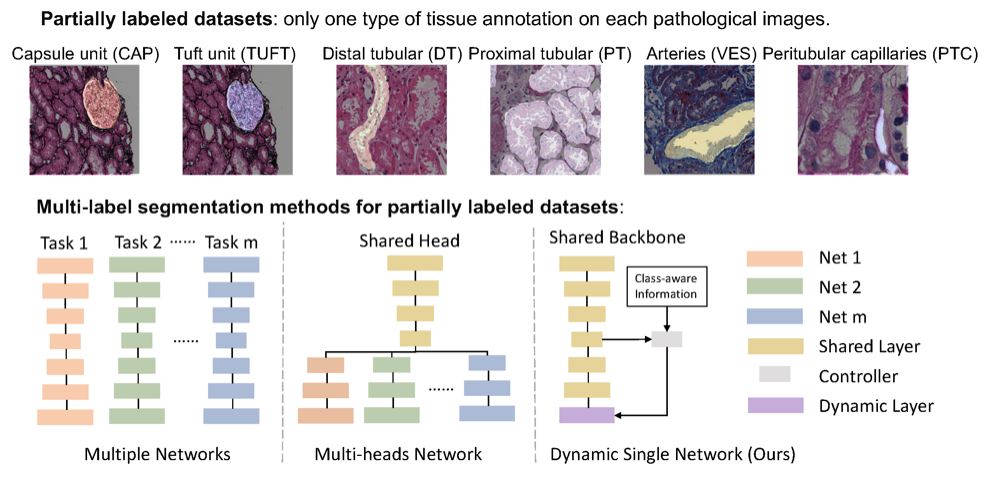 Single dynamic network for multi-label renal pathology image segmentationRuining Deng, Quan Liu , Can Cui , Zuhayr Asad , Haichun Yang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning , 2022
Single dynamic network for multi-label renal pathology image segmentationRuining Deng, Quan Liu , Can Cui , Zuhayr Asad , Haichun Yang , and Yuankai HuoIn International Conference on Medical Imaging with Deep Learning , 2022Computer-assisted quantitative analysis on Giga-pixel pathology images has provided a new avenue in histology examination. The innovations have been largely focused on cancer pathology (i.e., tumor segmentation and characterization). In non-cancer pathology, the learning algorithms can be asked to examine more comprehensive tissue types simultaneously, as a multi-label setting. The prior arts typically needed to train multiple segmentation networks in order to match the domain-specific knowledge for heterogeneous tissue types (e.g., glomerular tuft, glomerular unit, proximal tubular, distal tubular, peritubular capillaries, and arteries). In this paper, we propose a dynamic single segmentation network (Omni-Seg) that learns to segment multiple tissue types using partially labeled images (i.e., only one tissue type is labeled for each training image) for renal pathology. By learning from 150,000 patch-wise pathological images from six tissue types, the proposed Omni-Seg network achieved superior segmentation accuracy and less resource consumption when compared to the previous the multiple-network and multi-head design. In the testing stage, the proposed method obtains “completely labeled" tissue segmentation results using only “partially labeled" training images. The source code is available at \urlhttps://github.com/ddrrnn123/Omni-Seg
@inproceedings{deng2022single, title = {Single dynamic network for multi-label renal pathology image segmentation}, author = {Deng, Ruining and Liu, Quan and Cui, Can and Asad, Zuhayr and Yang, Haichun and Huo, Yuankai}, booktitle = {International Conference on Medical Imaging with Deep Learning}, pages = {304--314}, year = {2022}, organization = {PMLR}, } -
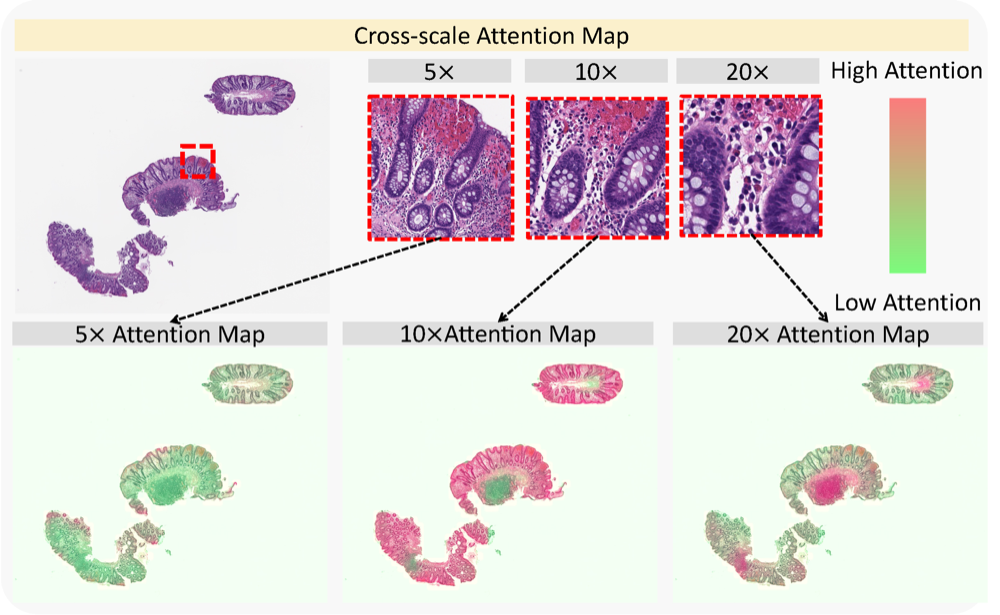 Cross-scale attention guided multi-instance learning for Crohn’s disease diagnosis with pathological imagesRuining Deng, Can Cui , Lucas W Remedios , Shunxing Bao , R Michael Womick , Sophie Chiron , Jia Li , Joseph T Roland , Ken S Lau , Qi Liu , Keith T Wilson , Yaohong Wang , Lori A Coburn , Bennett A Landman , and Yuankai HuoIn International Workshop on Multiscale Multimodal Medical Imaging , 2022
Cross-scale attention guided multi-instance learning for Crohn’s disease diagnosis with pathological imagesRuining Deng, Can Cui , Lucas W Remedios , Shunxing Bao , R Michael Womick , Sophie Chiron , Jia Li , Joseph T Roland , Ken S Lau , Qi Liu , Keith T Wilson , Yaohong Wang , Lori A Coburn , Bennett A Landman , and Yuankai HuoIn International Workshop on Multiscale Multimodal Medical Imaging , 2022Multi-instance learning (MIL) is widely used in the computer-aided interpretation of pathological Whole Slide Images (WSIs) to solve the lack of pixel-wise or patch-wise annotations. Often, this approach directly applies “natural image driven” MIL algorithms which overlook the multi-scale (i.e. pyramidal) nature of WSIs. Off-the-shelf MIL algorithms are typically deployed on a single-scale of WSIs (e.g., 20 magnification), while human pathologists usually aggregate the global and local patterns in a multi-scale manner (e.g., by zooming in and out between different magnifications). In this study, we propose a novel cross-scale attention mechanism to explicitly aggregate inter-scale interactions into a single MIL network for Crohn’s Disease (CD), which is a form of inflammatory bowel disease. The contribution of this paper is two-fold: (1) a cross-scale attention mechanism is proposed to aggregate features from different resolutions with multi-scale interaction; and (2) differential multi-scale attention visualizations are generated to localize explainable lesion patterns. By training 250,000 H &E-stained Ascending Colon (AC) patches from 20 CD patient and 30 healthy control samples at different scales, our approach achieved a superior Area under the Curve (AUC) score of 0.8924 compared with baseline models. The official implementation is publicly available at https://github.com/hrlblab/CS-MIL.
@inproceedings{deng2022cross, title = {Cross-scale attention guided multi-instance learning for Crohn’s disease diagnosis with pathological images}, author = {Deng, Ruining and Cui, Can and Remedios, Lucas W and Bao, Shunxing and Womick, R Michael and Chiron, Sophie and Li, Jia and Roland, Joseph T and Lau, Ken S and Liu, Qi and Wilson, Keith T and Wang, Yaohong and Coburn, Lori A and Landman, Bennett A and Huo, Yuankai}, booktitle = {International Workshop on Multiscale Multimodal Medical Imaging}, pages = {24--33}, year = {2022}, organization = {Springer}, } -
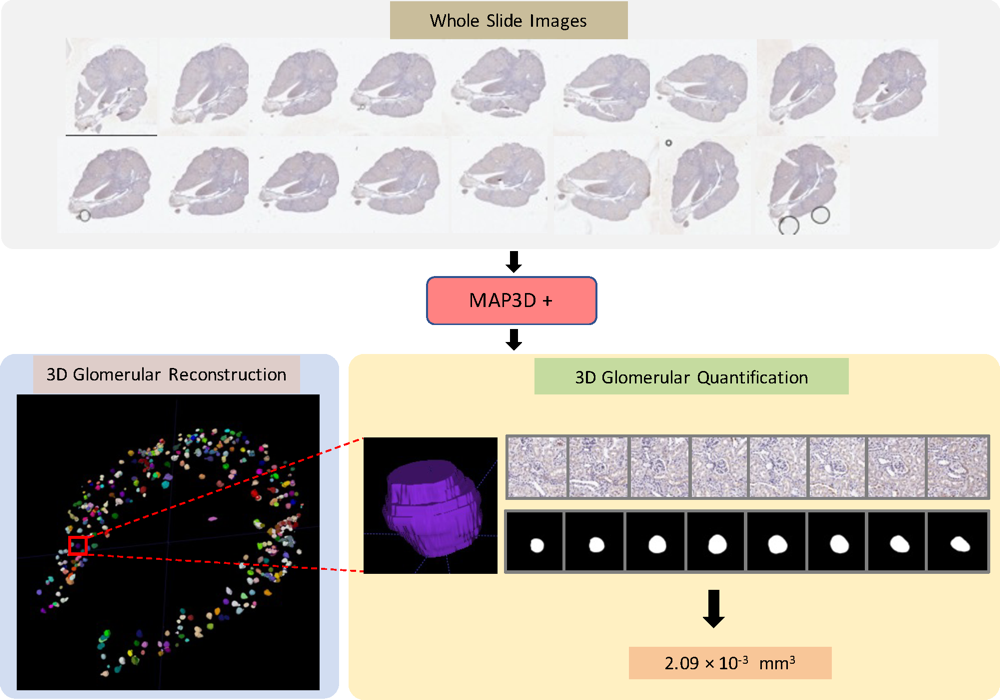 Dense multi-object 3D glomerular reconstruction and quantification on 2D serial section whole slide imagesRuining Deng, Haichun Yang , Zuhayr Asad , Zheyu Zhu , Shiru Wang , Lee E Wheless , Agnes B Fogo , and Yuankai HuoIn Medical Imaging 2022: Digital and Computational Pathology , 2022
Dense multi-object 3D glomerular reconstruction and quantification on 2D serial section whole slide imagesRuining Deng, Haichun Yang , Zuhayr Asad , Zheyu Zhu , Shiru Wang , Lee E Wheless , Agnes B Fogo , and Yuankai HuoIn Medical Imaging 2022: Digital and Computational Pathology , 2022There has been a long pursuit for precise and reproducible glomerular quantification in the field of renal pathology in both research and clinical practice. Currently, 3D glomerular identification and reconstruction of large-scale glomeruli are labor-intensive tasks, and time-consuming by manual analysis on whole slide imaging (WSI) in 2D serial sectioning representation. The accuracy of serial section analysis is also limited in the 2D serial context. Moreover, there are no approaches to present 3D glomerular visualization for human examination (volume calculation, 3D phenotype analysis, etc.). In this paper, we introduce an end-to-end holistic deep-learning-based method that achieves automatic detection, segmentation and multi-object tracking (MOT) of individual glomeruli with large-scale glomerular-registered assessment in a 3D context on WSIs. The high-resolution WSIs are the inputs, while the outputs are the 3D glomerular reconstruction and volume estimation. This pipeline achieves 81.8 in IDF1 and 69.1 in MOTA as MOT performance, while the proposed volume estimation achieves 0.84 Spearman correlation coefficient with manual annotation. The end-to-end MAP3D+ pipeline provides an approach for extensive 3D glomerular reconstruction and volume quantification from 2D serial section WSIs.
@inproceedings{deng2022dense, title = {Dense multi-object 3D glomerular reconstruction and quantification on 2D serial section whole slide images}, author = {Deng, Ruining and Yang, Haichun and Asad, Zuhayr and Zhu, Zheyu and Wang, Shiru and Wheless, Lee E and Fogo, Agnes B and Huo, Yuankai}, booktitle = {Medical Imaging 2022: Digital and Computational Pathology}, volume = {12039}, pages = {83--90}, year = {2022}, organization = {SPIE}, }
2021
-
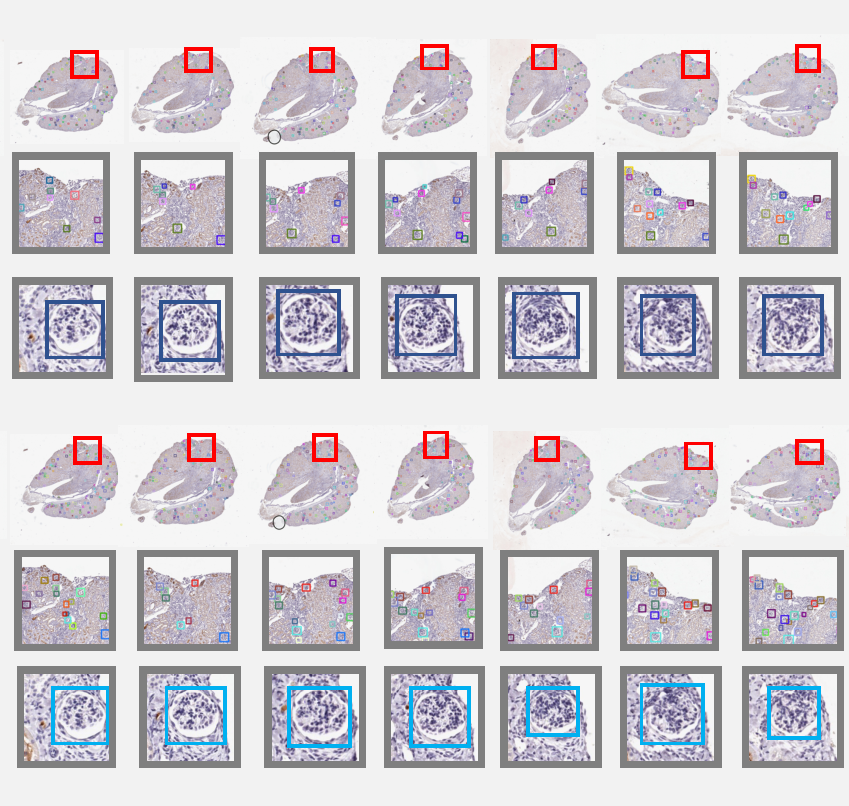 Map3d: registration-based multi-object tracking on 3d serial whole slide imagesRuining Deng, Haichun Yang , Aadarsh Jha , Yuzhe Lu , Peng Chu , Agnes B Fogo , and Yuankai HuoIEEE transactions on medical imaging, 2021
Map3d: registration-based multi-object tracking on 3d serial whole slide imagesRuining Deng, Haichun Yang , Aadarsh Jha , Yuzhe Lu , Peng Chu , Agnes B Fogo , and Yuankai HuoIEEE transactions on medical imaging, 2021There has been a long pursuit for precise and reproducible glomerular quantification on renal pathology to leverage both research and practice. When digitizing the biopsy tissue samples using whole slide imaging (WSI), a set of serial sections from the same tissue can be acquired as a stack of images, similar to frames in a video. In radiology, the stack of images (e.g., computed tomography) are naturally used to provide 3D context for organs, tissues, and tumors. In pathology, it is appealing to do a similar 3D assessment. However, the 3D identification and association of large-scale glomeruli on renal pathology is challenging due to large tissue deformation, missing tissues, and artifacts from WSI. In this paper, we propose a novel Multi-object Association for Pathology in 3D (Map3D) method for automatically identifying and associating large-scale cross-sections of 3D objects from routine serial sectioning and WSI. The innovations of the Multi-Object Association for Pathology in 3D (Map3D) method are three-fold: (1) the large-scale glomerular association is formed as a new multi-object tracking (MOT) perspective; (2) the quality-aware whole series registration is proposed to not only provide affinity estimation but also offer automatic kidney-wise quality assurance (QA) for registration; (3) a dual-path association method is proposed to tackle the large deformation, missing tissues, and artifacts during tracking. To the best of our knowledge, the Map3D method is the first approach that enables automatic and large-scale glomerular association across 3D serial sectioning using WSI. Our proposed method Map3D achieved MOTA = 44.6, which is 12.1% higher than the non-deep learning benchmarks.
@article{deng2021map3d, title = {Map3d: registration-based multi-object tracking on 3d serial whole slide images}, author = {Deng, Ruining and Yang, Haichun and Jha, Aadarsh and Lu, Yuzhe and Chu, Peng and Fogo, Agnes B and Huo, Yuankai}, journal = {IEEE transactions on medical imaging}, volume = {40}, number = {7}, pages = {1924--1933}, year = {2021}, publisher = {IEEE}, } -
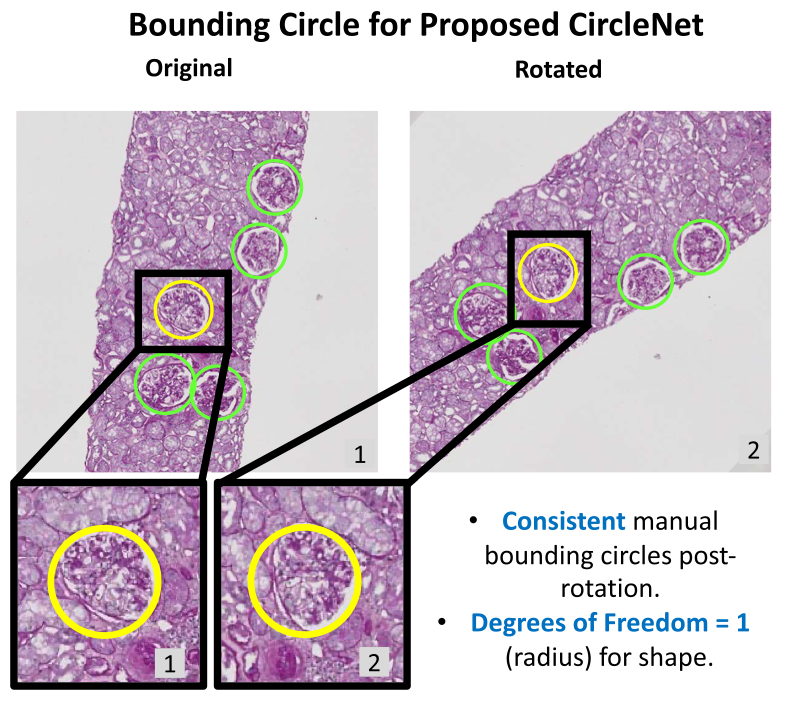 Circle representation for medical object detectionEthan H Nguyen , Haichun Yang , Ruining Deng, Yuzhe Lu , Zheyu Zhu , Joseph T Roland , Le Lu , Bennett A Landman , Agnes B Fogo , and Yuankai HuoIEEE transactions on medical imaging, 2021
Circle representation for medical object detectionEthan H Nguyen , Haichun Yang , Ruining Deng, Yuzhe Lu , Zheyu Zhu , Joseph T Roland , Le Lu , Bennett A Landman , Agnes B Fogo , and Yuankai HuoIEEE transactions on medical imaging, 2021Box representation has been extensively used for object detection in computer vision. Such representation is efficacious but not necessarily optimized for biomedical objects (e.g., glomeruli), which play an essential role in renal pathology. In this paper, we propose a simple circle representation for medical object detection and introduce CircleNet, an anchor-free detection framework. Compared with the conventional bounding box representation, the proposed bounding circle representation innovates in three-fold: (1) it is optimized for ball-shaped biomedical objects; (2) The circle representation reduced the degree of freedom compared with box representation; (3) It is naturally more rotation invariant. When detecting glomeruli and nuclei on pathological images, the proposed circle representation achieved superior detection performance and be more rotation-invariant, compared with the bounding box. The code has been made publicly available: https://github.com/hrlblab/CircleNet.
@article{nguyen2021circle, title = {Circle representation for medical object detection}, author = {Nguyen, Ethan H and Yang, Haichun and Deng, Ruining and Lu, Yuzhe and Zhu, Zheyu and Roland, Joseph T and Lu, Le and Landman, Bennett A and Fogo, Agnes B and Huo, Yuankai}, journal = {IEEE transactions on medical imaging}, volume = {41}, number = {3}, pages = {746--754}, year = {2021}, publisher = {IEEE}, } -
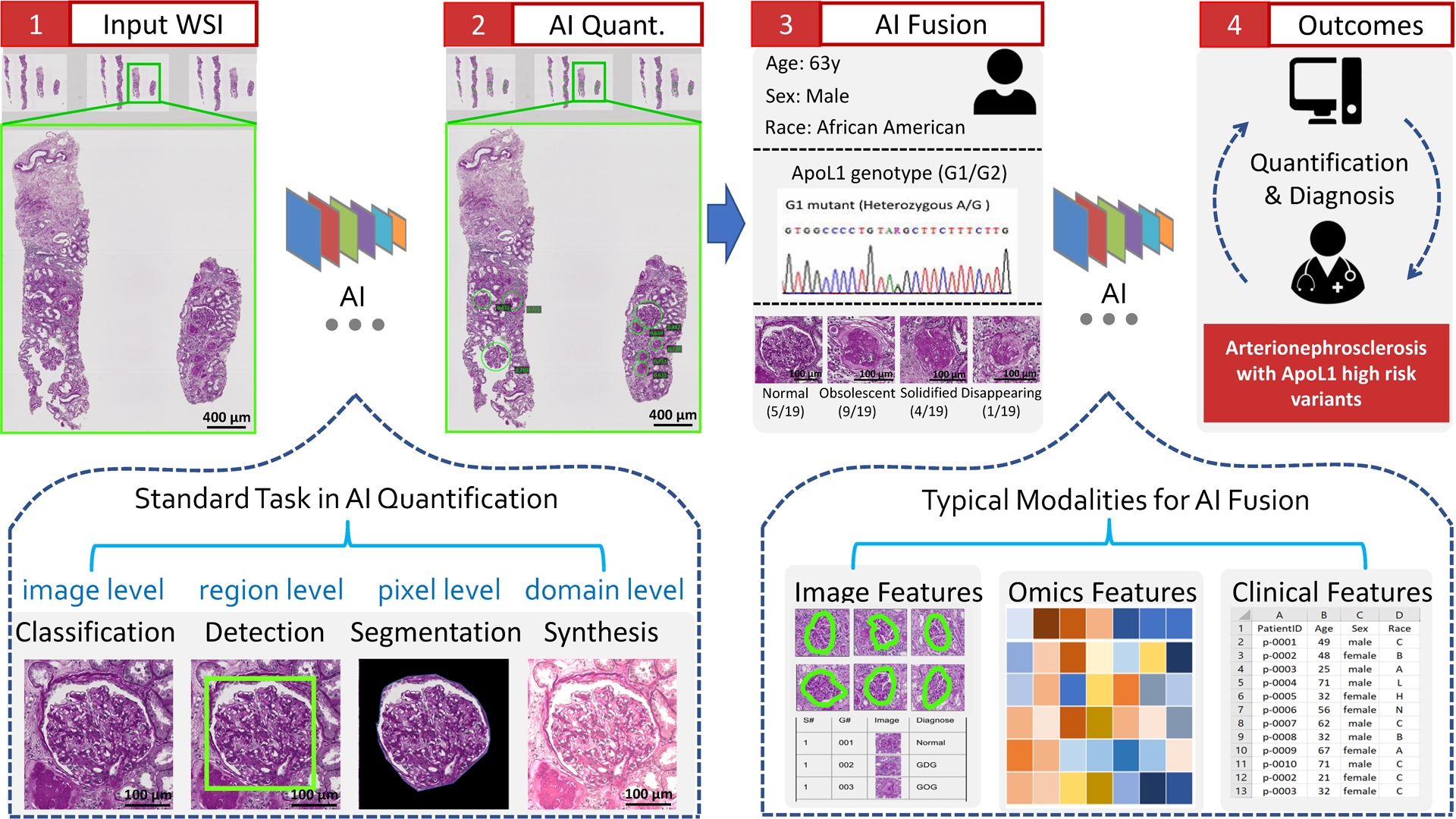 AI applications in renal pathologyYuankai Huo , Ruining Deng, Quan Liu , Agnes B Fogo , and Haichun YangKidney international, 2021
AI applications in renal pathologyYuankai Huo , Ruining Deng, Quan Liu , Agnes B Fogo , and Haichun YangKidney international, 2021The explosive growth of artificial intelligence (AI) technologies, especially deep learning methods, has been translated at revolutionary speed to efforts in AI-assisted healthcare. New applications of AI to renal pathology have recently become available, driven by the successful AI deployments in digital pathology. However, synergetic developments of renal pathology and AI require close interdisciplinary collaborations between computer scientists and renal pathologists. Computer scientists should understand that not every AI innovation is translatable to renal pathology, while renal pathologists should capture high-level principles of the relevant AI technologies. Herein, we provide an integrated review on current and possible future applications in AI-assisted renal pathology, by including perspectives from computer scientists and renal pathologists. First, the standard stages, from data collection to analysis, in full-stack AI-assisted renal pathology studies are reviewed. Second, representative renal pathology-optimized AI techniques are introduced. Last, we review current clinical AI applications, as well as promising future applications with the recent advances in AI.
@article{huo2021ai, title = {AI applications in renal pathology}, author = {Huo, Yuankai and Deng, Ruining and Liu, Quan and Fogo, Agnes B and Yang, Haichun}, journal = {Kidney international}, volume = {99}, number = {6}, pages = {1309--1320}, year = {2021}, publisher = {Elsevier}, } -
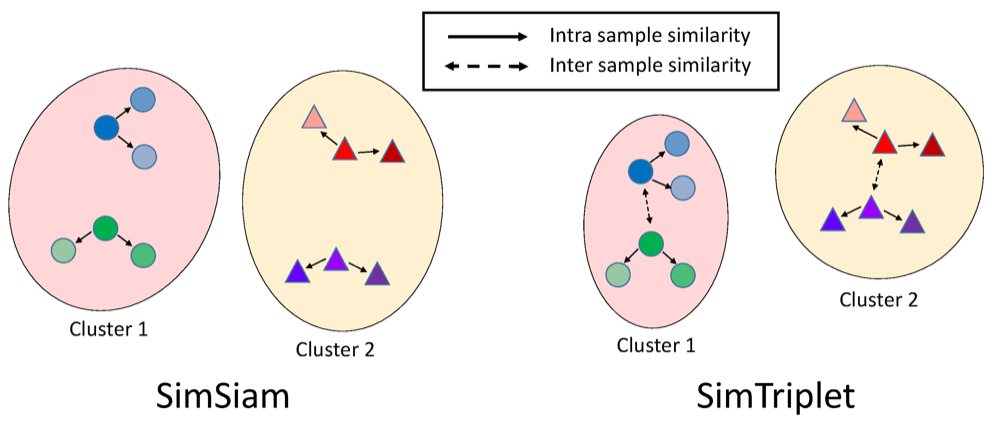 Simtriplet: Simple triplet representation learning with a single gpuQuan Liu , Peter C Louis , Yuzhe Lu , Aadarsh Jha , Mengyang Zhao , Ruining Deng, Tianyuan Yao , Joseph T Roland , Haichun Yang , Shilin Zhao , Lee E Wheless , and Yuankai HuoIn Medical Image Computing and Computer Assisted Intervention–MICCAI 2021: 24th International Conference, Strasbourg, France, September 27–October 1, 2021, Proceedings, Part II 24 , 2021
Simtriplet: Simple triplet representation learning with a single gpuQuan Liu , Peter C Louis , Yuzhe Lu , Aadarsh Jha , Mengyang Zhao , Ruining Deng, Tianyuan Yao , Joseph T Roland , Haichun Yang , Shilin Zhao , Lee E Wheless , and Yuankai HuoIn Medical Image Computing and Computer Assisted Intervention–MICCAI 2021: 24th International Conference, Strasbourg, France, September 27–October 1, 2021, Proceedings, Part II 24 , 2021Contrastive learning is a key technique of modern self-supervised learning. The broader accessibility of earlier approaches is hindered by the need of heavy computational resources (e.g., at least 8 GPUs or 32 TPU cores), which accommodate for large-scale negative samples or momentum. The more recent SimSiam approach addresses such key limitations via stop-gradient without momentum encoders. In medical image analysis, multiple instances can be achieved from the same patient or tissue. Inspired by these advances, we propose a simple triplet representation learning (SimTriplet) approach on pathological images. The contribution of the paper is three-fold: (1) The proposed SimTriplet method takes advantage of the multi-view nature of medical images beyond self-augmentation; (2) The method maximizes both intra-sample and inter-sample similarities via triplets from positive pairs, without using negative samples; and (3) The recent mix precision training is employed to advance the training by only using a single GPU with 16 GB memory. By learning from 79,000 unlabeled pathological patch images, SimTriplet achieved 10.58% better performance compared with supervised learning. It also achieved 2.13% better performance compared with SimSiam. Our proposed SimTriplet can achieve decent performance using only 1% labeled data. The code and data are available at https://github.com/hrlblab/SimTriplet.
@inproceedings{liu2021simtriplet, title = {Simtriplet: Simple triplet representation learning with a single gpu}, author = {Liu, Quan and Louis, Peter C and Lu, Yuzhe and Jha, Aadarsh and Zhao, Mengyang and Deng, Ruining and Yao, Tianyuan and Roland, Joseph T and Yang, Haichun and Zhao, Shilin and Wheless, Lee E and Huo, Yuankai}, booktitle = {Medical Image Computing and Computer Assisted Intervention--MICCAI 2021: 24th International Conference, Strasbourg, France, September 27--October 1, 2021, Proceedings, Part II 24}, pages = {102--112}, year = {2021}, organization = {Springer}, } -
 CaCL: Class-Aware Codebook Learning for Weakly Supervised Segmentation on Diffuse Image PatternsRuining Deng, Quan Liu , Shunxing Bao , Aadarsh Jha , Catie Chang , Bryan A Millis , Matthew J Tyska , and Yuankai HuoIn Deep Generative Models, and Data Augmentation, Labelling, and Imperfections: First Workshop, DGM4MICCAI 2021, and First Workshop, DALI 2021, Held in Conjunction with MICCAI 2021, Strasbourg, France, October 1, 2021, Proceedings 1 , 2021
CaCL: Class-Aware Codebook Learning for Weakly Supervised Segmentation on Diffuse Image PatternsRuining Deng, Quan Liu , Shunxing Bao , Aadarsh Jha , Catie Chang , Bryan A Millis , Matthew J Tyska , and Yuankai HuoIn Deep Generative Models, and Data Augmentation, Labelling, and Imperfections: First Workshop, DGM4MICCAI 2021, and First Workshop, DALI 2021, Held in Conjunction with MICCAI 2021, Strasbourg, France, October 1, 2021, Proceedings 1 , 2021Weakly supervised learning has been rapidly advanced in biomedical image analysis to achieve pixel-wise labels (segmentation) from image-wise annotations (classification), as biomedical images naturally contain image-wise labels in many scenarios. The current weakly supervised learning algorithms from the computer vision community are largely designed for focal objects (e.g., dogs and cats). However, such algorithms are not optimized for diffuse patterns in biomedical imaging (e.g., stains and fluorescence in microscopy imaging). In this paper, we propose a novel class-aware codebook learning (CaCL) algorithm to perform weakly supervised learning for diffuse image patterns. Specifically, the CaCL algorithm is deployed to segment protein expressed brush border regions from histological images of human duodenum. Our contribution is three-fold: (1) we approach the weakly supervised segmentation from a novel codebook learning perspective; (2) the CaCL algorithm segments diffuse image patterns rather than focal objects; and (3) the proposed algorithm is implemented in a multi-task framework based on Vector Quantised-Variational AutoEncoder (VQ-VAE) via joint image reconstruction, classification, feature embedding, and segmentation. The experimental results show that our method achieved superior performance compared with baseline weakly supervised algorithms. The code is available at https://github.com/ddrrnn123/CaCL.
@inproceedings{deng2021cacl, title = {CaCL: Class-Aware Codebook Learning for Weakly Supervised Segmentation on Diffuse Image Patterns}, author = {Deng, Ruining and Liu, Quan and Bao, Shunxing and Jha, Aadarsh and Chang, Catie and Millis, Bryan A and Tyska, Matthew J and Huo, Yuankai}, booktitle = {Deep Generative Models, and Data Augmentation, Labelling, and Imperfections: First Workshop, DGM4MICCAI 2021, and First Workshop, DALI 2021, Held in Conjunction with MICCAI 2021, Strasbourg, France, October 1, 2021, Proceedings 1}, pages = {93--102}, year = {2021}, organization = {Springer}, }
2020
-
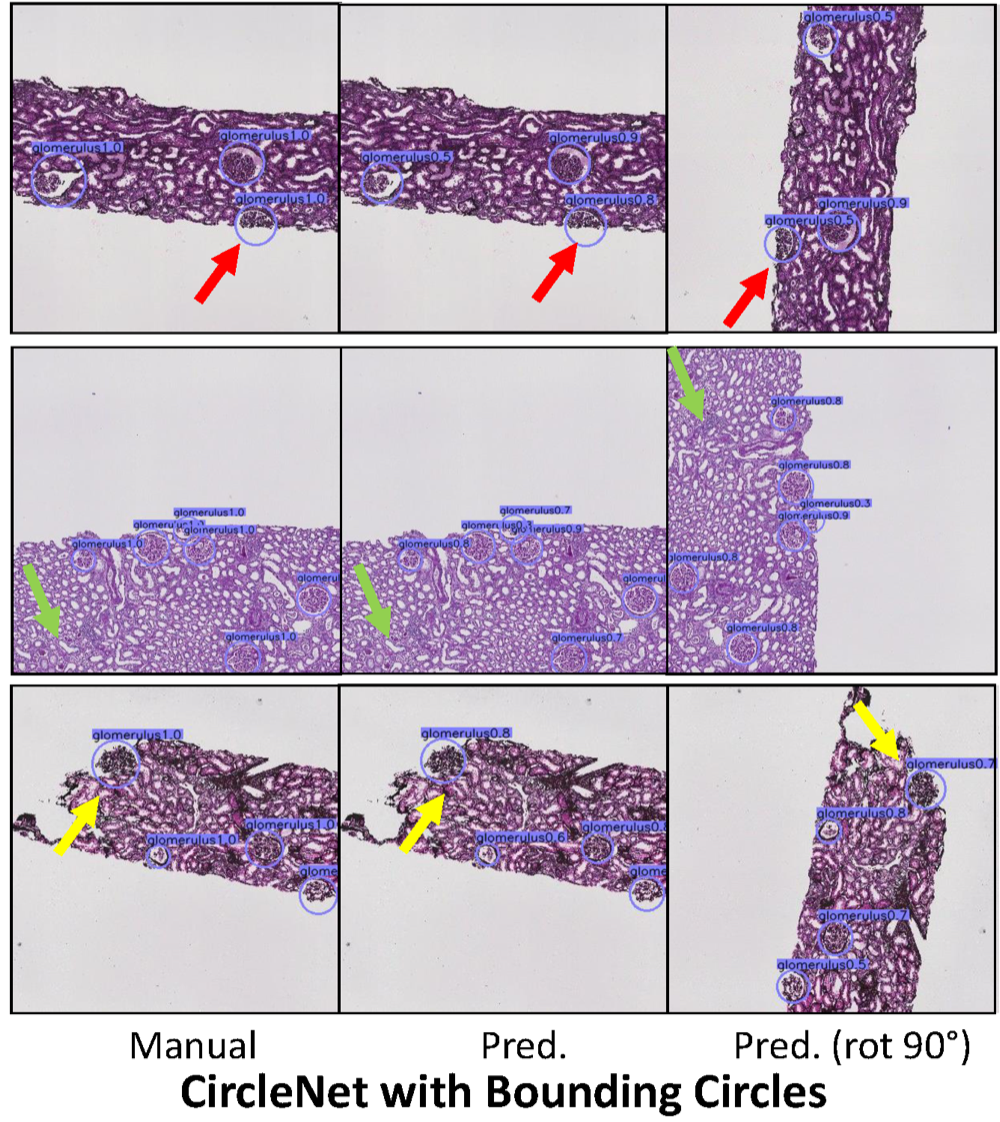 CircleNet: Anchor-free glomerulus detection with circle representationHaichun Yang , Ruining Deng, Yuzhe Lu , Zheyu Zhu , Ye Chen , Joseph T Roland , Le Lu , Bennett A Landman , Agnes B Fogo , and Yuankai HuoIn Medical Image Computing and Computer Assisted Intervention–MICCAI 2020: 23rd International Conference, Lima, Peru, October 4–8, 2020, Proceedings, Part IV 23 , 2020
CircleNet: Anchor-free glomerulus detection with circle representationHaichun Yang , Ruining Deng, Yuzhe Lu , Zheyu Zhu , Ye Chen , Joseph T Roland , Le Lu , Bennett A Landman , Agnes B Fogo , and Yuankai HuoIn Medical Image Computing and Computer Assisted Intervention–MICCAI 2020: 23rd International Conference, Lima, Peru, October 4–8, 2020, Proceedings, Part IV 23 , 2020Object detection networks are powerful in computer vision, but not necessarily optimized for biomedical object detection. In this work, we propose CircleNet, a simple anchor-free detection method with circle representation for detection of the ball-shaped glomerulus. Different from the traditional bounding box based detection method, the bounding circle (1) reduces the degrees of freedom of detection representation, (2) is naturally rotation invariant, (3) and optimized for ball-shaped objects. The key innovation to enable this representation is the anchor-free framework with the circle detection head. We evaluate CircleNet in the context of detection of glomerulus. CircleNet increases average precision of the glomerulus detection from 0.598 to 0.647. Another key advantage is that CircleNet achieves better rotation consistency compared with bounding box representations.
@inproceedings{yang2020circlenet, title = {CircleNet: Anchor-free glomerulus detection with circle representation}, author = {Yang, Haichun and Deng, Ruining and Lu, Yuzhe and Zhu, Zheyu and Chen, Ye and Roland, Joseph T and Lu, Le and Landman, Bennett A and Fogo, Agnes B and Huo, Yuankai}, booktitle = {Medical Image Computing and Computer Assisted Intervention--MICCAI 2020: 23rd International Conference, Lima, Peru, October 4--8, 2020, Proceedings, Part IV 23}, pages = {35--44}, year = {2020}, organization = {Springer}, }